

Soluble beta-amyloid1-40 induces NMDA-dependent degradation of postsynaptic density-95 at glutamatergic synapses
- PMID: 16319306
- PMCID: PMC6725651
- DOI: 10.1523/JNEUROSCI.3034-05.2005
Soluble beta-amyloid1-40 induces NMDA-dependent degradation of postsynaptic density-95 at glutamatergic synapses
Abstract
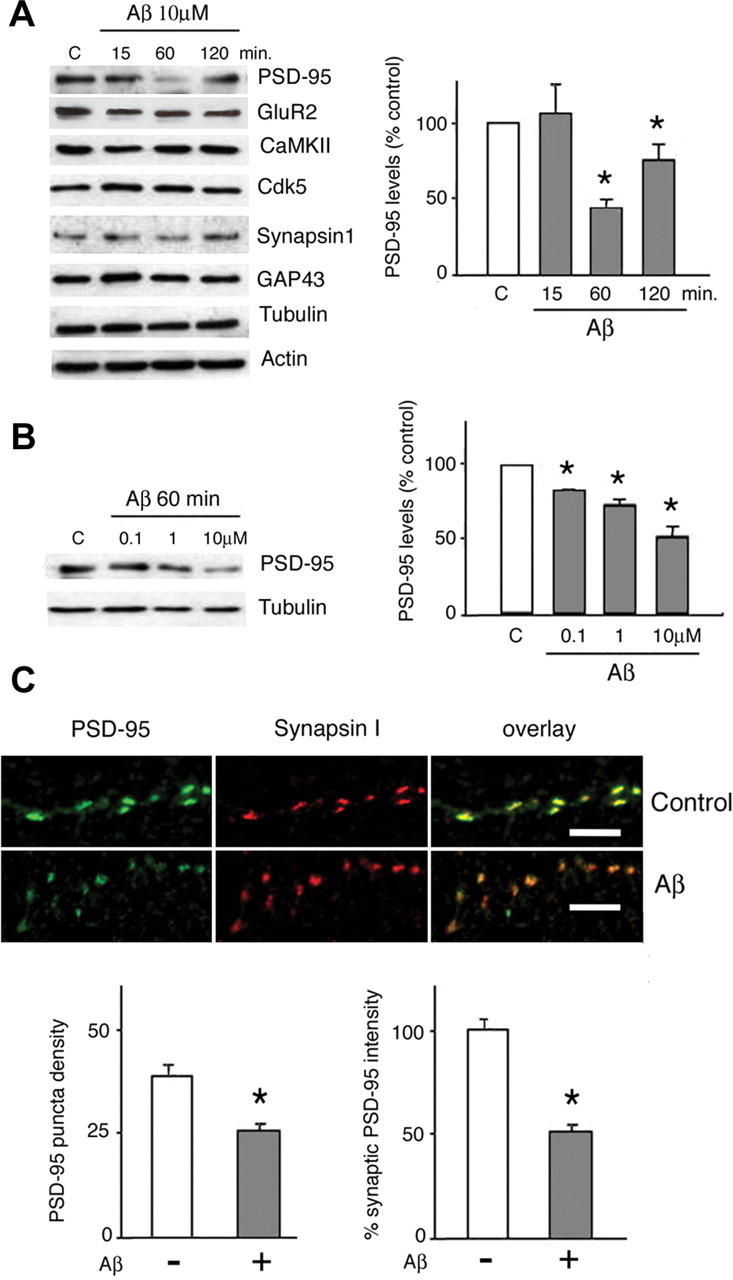
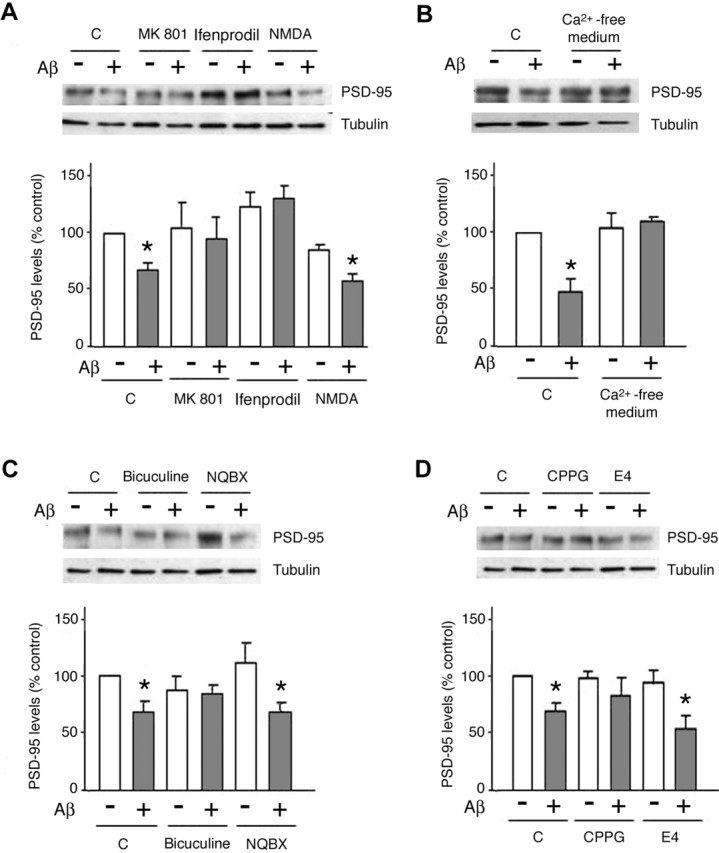
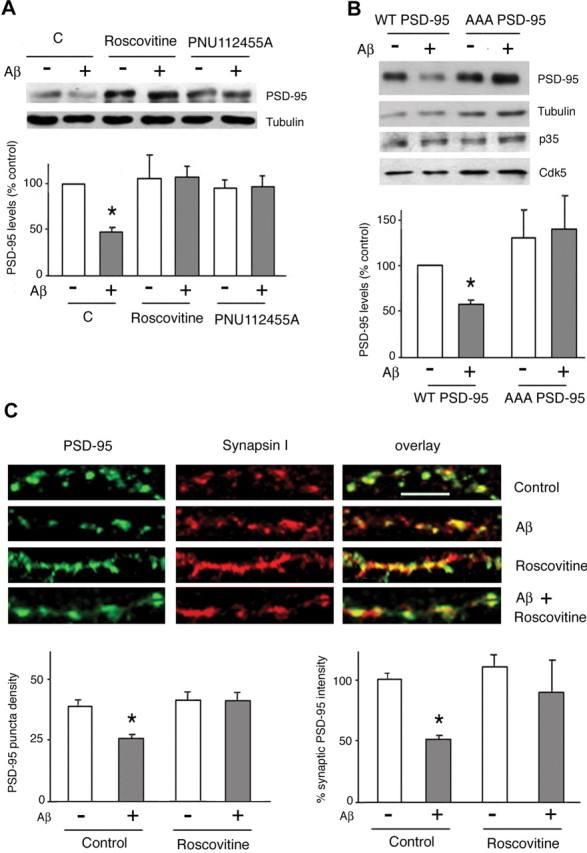
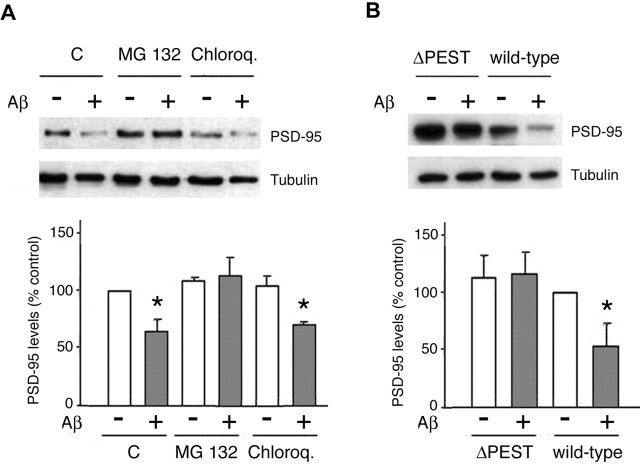
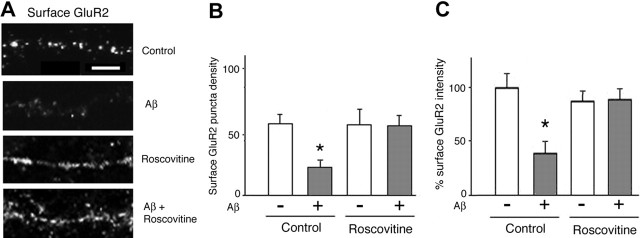
Amyloid-beta (Abeta) has been implicated in memory loss and disruption of synaptic plasticity observed in early-stage Alzheimer's disease. Recently, it has been shown that soluble Abeta oligomers target synapses in cultured rat hippocampal neurons, suggesting a direct role of Abeta in the regulation of synaptic structure and function. Postsynaptic density-95 (PSD-95) is a postsynaptic scaffolding protein that plays a critical role in synaptic plasticity and the stabilization of AMPA (AMPARs) and NMDA (NMDARs) receptors at synapses. Here, we show that exposure of cultured cortical neurons to soluble oligomers of Abeta(1-40) reduces PSD-95 protein levels in a dose- and time-dependent manner and that the Abeta1(1-40)-dependent decrease in PSD-95 requires NMDAR activity. We also show that the decrease in PSD-95 requires cyclin-dependent kinase 5 activity and involves the proteasome pathway. Immunostaining analysis of cortical cultured neurons revealed that Abeta treatment induces concomitant decreases in PSD-95 at synapses and in the surface expression of the AMPAR glutamate receptor subunit 2. Together, these data suggest a novel pathway by which Abeta triggers synaptic dysfunction, namely, by altering the molecular composition of glutamatergic synapses.
Figures
References
-
- Bingol B, Schuman E (2004) A proteasome-sensitive connection between PSD-95 and GluR1 endocytosis. Neuropharmacology 47: 755-763. - PubMed
-
- Bitan G, Lomakin A, Teplow DB (2001) Amyloid β protein oligomerization prenucleation interactions revealed by photo-induced cross-linking of unmodified proteins. J Biol Chem 276: 35176-35184. - PubMed
-
- Bitan G, Vollers SS, Teplow DB (2003) Elucidation of primary structure elements controlling early amyloid beta-protein oligomerization. J Biol Chem 278: 34882-34889. - PubMed
-
- Bredt D, Nicoll R (2003) AMPA receptor trafficking at excitatory synapses. Neuron 40: 361-379. - PubMed
-
- Chen L, Chetkovich DM, Petralia RS, Sweeney NT, Kawasaki Y, Wenthold RJ, Bredt DS, Nicoll RA (2000) Stargazin regulates synaptic targeting of AMPA receptors by two distinct mechanisms. Nature 408: 936-943. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources