

The role of an astrocytic NADPH oxidase in the neurotoxicity of amyloid beta peptides
- PMID: 16321801
- PMCID: PMC1569597
- DOI: 10.1098/rstb.2005.1766
The role of an astrocytic NADPH oxidase in the neurotoxicity of amyloid beta peptides
Abstract
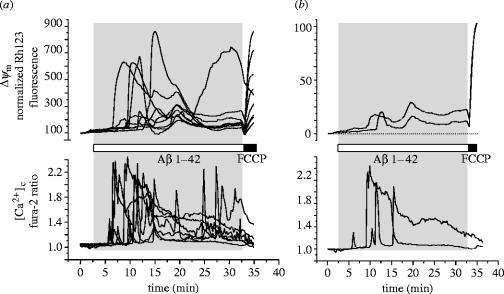
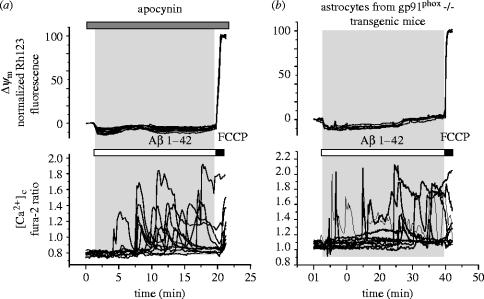
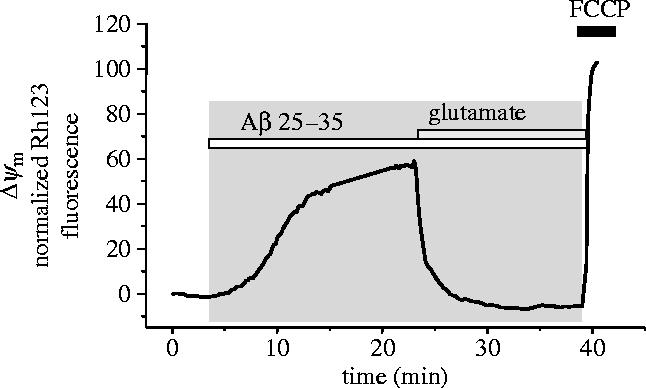
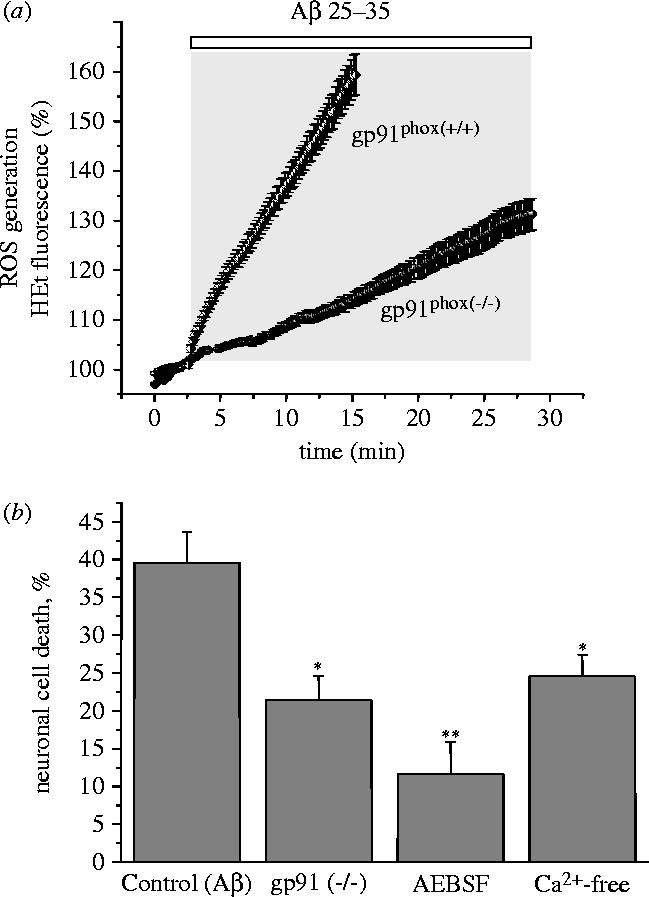
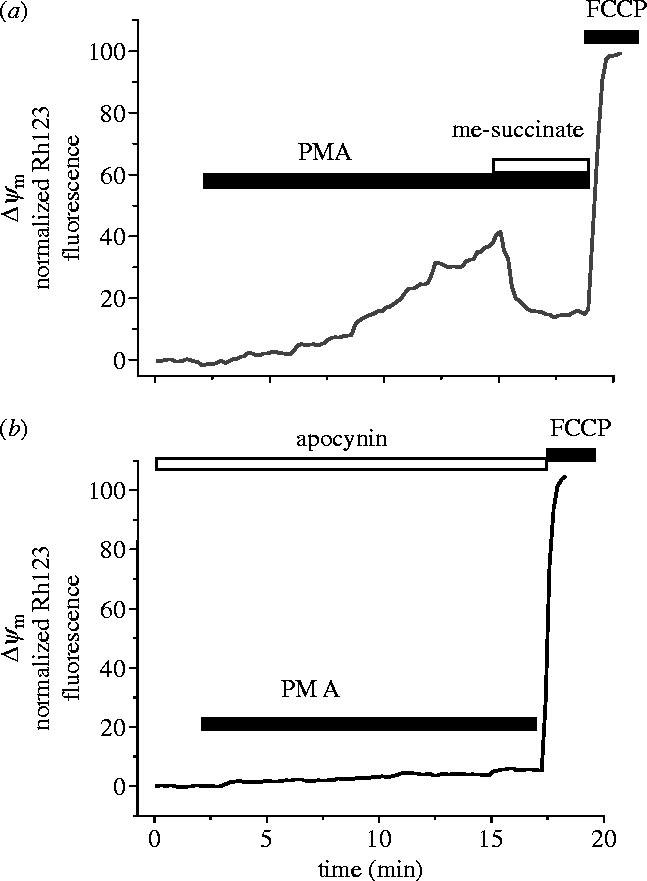
Amyloid beta peptide (Abeta) accumulates in the CNS in Alzheimer's disease. Both the full peptide (1-42) or the 25-35 fragment are toxic to neurons in culture. We have used fluorescence imaging technology to explore the mechanism of neurotoxicity in mixed asytrocyte/neuronal cultures prepared from rat or mouse cortex or hippocampus, and have found that Abeta acts preferentially on astrocytes but causes neuronal death. Abeta causes sporadic transient increases in [Ca2+]c in astrocytes, associated with a calcium dependent increased generation of reactive oxygen species (ROS) and glutathione depletion. This caused a slow dissipation of mitochondrial potential on which abrupt calcium dependent transient depolarizations were superimposed. The mitochondrial depolarization was reversed by mitochondrial substrates glutamate, pyruvate or methyl succinate, and by NADPH oxidase (NOX) inhibitors, suggesting that it reflects oxidative damage to metabolic pathways upstream of mitochondrial complex I. The Abeta induced increase in ROS and the mitochondrial depolarization were absent in cells cultured from transgenic mice lacking the NOX component, gp91phox. Neuronal death after 24 h of Abeta exposure was dramatically reduced both by NOX inhibitors and in gp91phox knockout mice. Thus, by raising [Ca2+]c in astrocytes, Abeta activates NOX, generating oxidative stress that is transmitted to neurons, causing neuronal death.
Figures
References
-
- Abramov A.Y, Canevari L, Duchen M.R. Calcium signals induced by amyloid beta peptide and their consequences in neurons and astrocytes in culture. Biochim. Biophys. Acta. 2004b;6:81–87. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous