

Changes in the chemical and dynamic properties of cardiac troponin T cause discrete cardiomyopathies in transgenic mice
- PMID: 16326803
- PMCID: PMC1298915
- DOI: 10.1073/pnas.0509181102
Changes in the chemical and dynamic properties of cardiac troponin T cause discrete cardiomyopathies in transgenic mice
Abstract
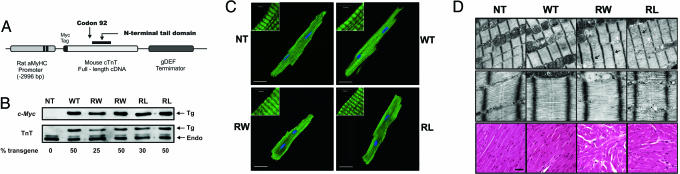
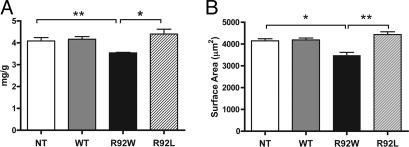
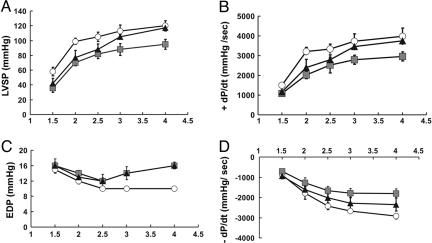
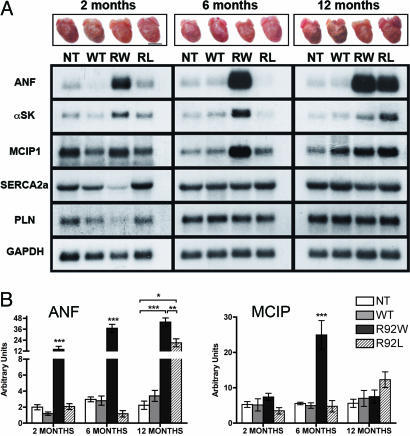
Cardiac troponin T (cTnT) is a central component of the regulatory thin filament. Mutations in cTnT have been linked to severe forms of familial hypertrophic cardiomyopathy. A mutational "hotspot" that leads to distinct clinical phenotypes has been identified at codon 92. Although the basic functional and structural roles of cTnT in modulating contractility are relatively well understood, the mechanisms that link point mutations in cTnT to the development of this complex cardiomyopathy are unknown. To address this question, we have taken a highly interdisciplinary approach by first determining the effects of the residue 92 mutations on the molecular flexibility and stability of cTnT by means of molecular dynamics simulations. To test whether the predicted alterations in thin filament structure could lead to distinct cardiomyopathies in vivo, we developed transgenic mouse models expressing either the Arg-92-Trp or Arg-92-Leu cTnT proteins in the heart. Characterization of these models at the cellular and whole-heart levels has revealed mutation-specific early alterations in transcriptional activation that result in distinct pathways of ventricular remodeling and contractile performance. Thus, our computational and experimental results show that changes in thin filament structure caused by single amino acid substitutions lead to differences in the biophysical properties of cTnT and alter disease pathogenesis.
Figures

References
-
- Tobacman, L. S. (1996) Annu. Rev. Physiol. 58, 447-481. - PubMed
-
- Li, M. X., Wang, X. & Sykes, B. D. (2004) J. Muscle Res. Cell Motil. 25, 559-579. - PubMed
-
- Watkins, H., McKenna, W. J., Thierfelder, L., Suk, H. J., Anan, R., O'Donoghue, A., Spirito, P., Matsumori, A., Moravec, C. S., Seidman, J. G. & Seidman, C. E. (1995) N. Engl. J. Med. 332, 1058-1064. - PubMed
-
- Forissier, J. F., Carrier, L., Farza, H., Bonne, G., Bercovici, J., Richard, P., Hainque, B., Townsend, P. J., Yacoub, M. H., Faure, S., et al. (1996) Circulation 94, 3069-3073. - PubMed
-
- Elliott, P. M., Sharma, S., Varnava, A., Poloniecki, J., Rowland, E. & McKenna, W. J. (1999) J. Am. Coll. Cardiol. 33, 1596-1601. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
