

Human plasma N-glycoproteome analysis by immunoaffinity subtraction, hydrazide chemistry, and mass spectrometry
- PMID: 16335952
- PMCID: PMC1850943
- DOI: 10.1021/pr0502065
Human plasma N-glycoproteome analysis by immunoaffinity subtraction, hydrazide chemistry, and mass spectrometry
Abstract
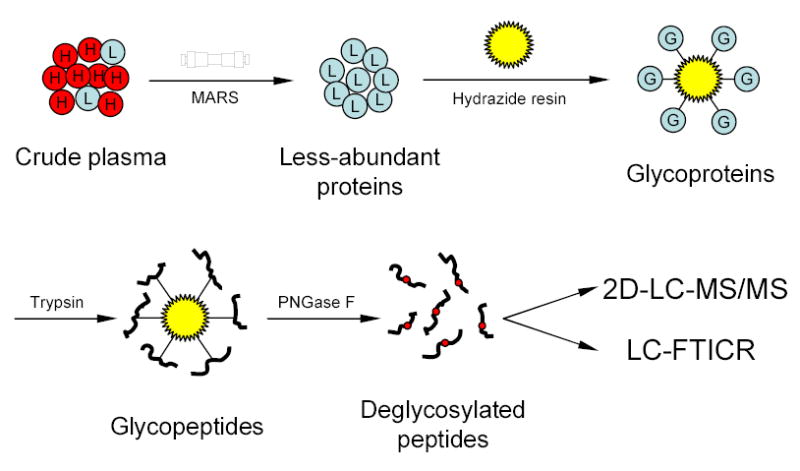
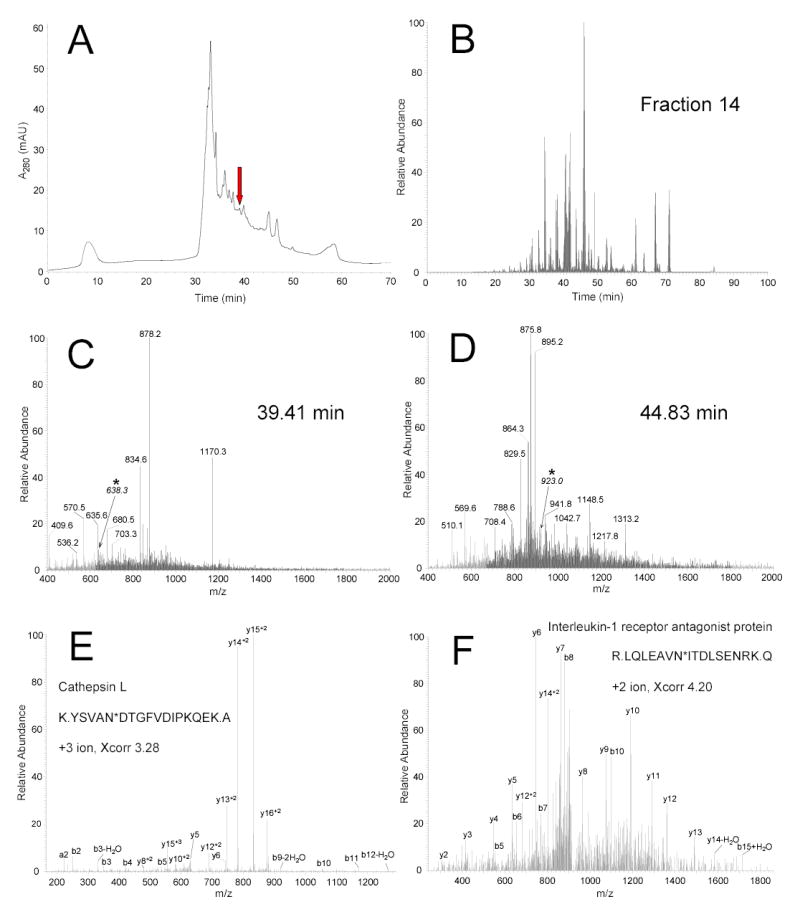
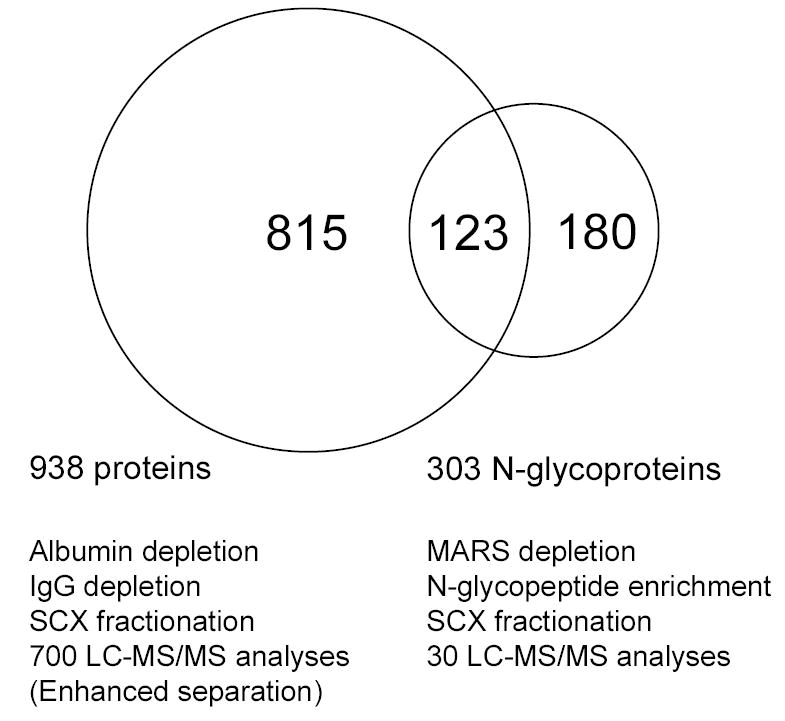
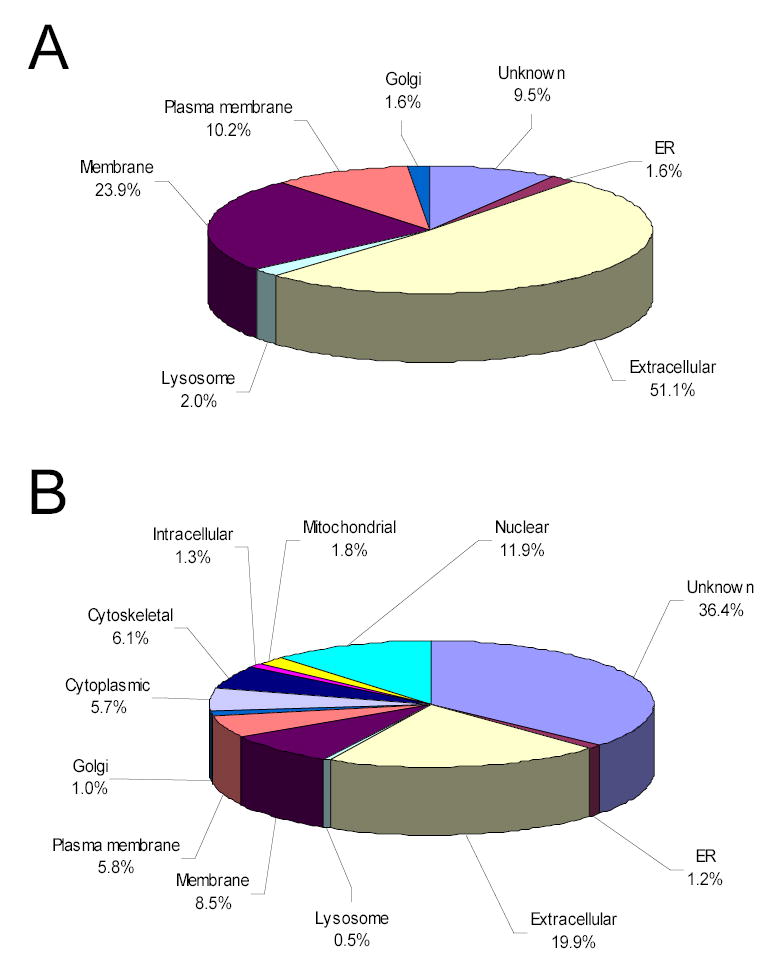
The enormous complexity, wide dynamic range of relative protein abundances of interest (over 10 orders of magnitude), and tremendous heterogeneity (due to post-translational modifications, such as glycosylation) of the human blood plasma proteome severely challenge the capabilities of existing analytical methodologies. Here, we describe an approach for broad analysis of human plasma N-glycoproteins using a combination of immunoaffinity subtraction and glycoprotein capture to reduce both the protein concentration range and the overall sample complexity. Six high-abundance plasma proteins were simultaneously removed using a pre-packed, immobilized antibody column. N-linked glycoproteins were then captured from the depleted plasma using hydrazide resin and enzymatically digested, and the bound N-linked glycopeptides were released using peptide-N-glycosidase F (PNGase F). Following strong cation exchange (SCX) fractionation, the deglycosylated peptides were analyzed by reversed-phase capillary liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS). Using stringent criteria, a total of 2053 different N-glycopeptides were confidently identified, covering 303 nonredundant N-glycoproteins. This enrichment strategy significantly improved detection and enabled identification of a number of low-abundance proteins, exemplified by interleukin-1 receptor antagonist protein (approximately 200 pg/mL), cathepsin L (approximately 1 ng/mL), and transforming growth factor beta 1 (approximately 2 ng/mL). A total of 639 N-glycosylation sites were identified, and the overall high accuracy of these glycosylation site assignments as assessed by accurate mass measurement using high-resolution liquid chromatography coupled to Fourier transform ion cyclotron resonance mass spectrometry (LC-FTICR) is initially demonstrated.
Figures
References
-
- Anderson NL, Anderson NG. Mol Cell Proteomics. 2002;1:845–867. - PubMed
-
- Diamandis EP. Mol Cell Proteomics. 2004;3:367–378. - PubMed
-
- Hanash S. Nature. 2003;422:226–232. - PubMed
-
- Petricoin E, Wulfkuhle J, Espina V, Liotta LA. J Proteome Res. 2004;3:209–217. - PubMed
-
- Pieper R, Gatlin CL, Makusky AJ, Russo PS, Schatz CR, Miller SS, Su Q, McGrath AM, Estock MA, Parmar PP, Zhao M, Huang ST, Zhou J, Wang F, Esquer-Blasco R, Anderson NL, Taylor J, Steiner S. Proteomics. 2003;3:1345–1364. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
