

Class II phosphoinositide 3-kinase alpha-isoform regulates Rho, myosin phosphatase and contraction in vascular smooth muscle
- PMID: 16336212
- PMCID: PMC1383708
- DOI: 10.1042/BJ20051471
Class II phosphoinositide 3-kinase alpha-isoform regulates Rho, myosin phosphatase and contraction in vascular smooth muscle
Abstract
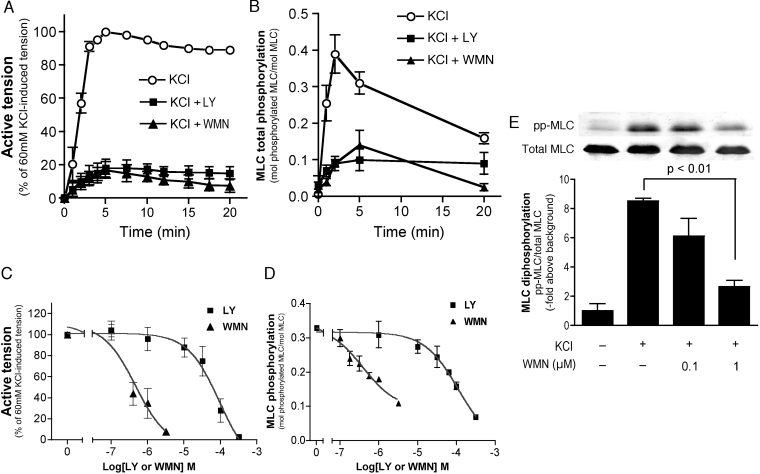
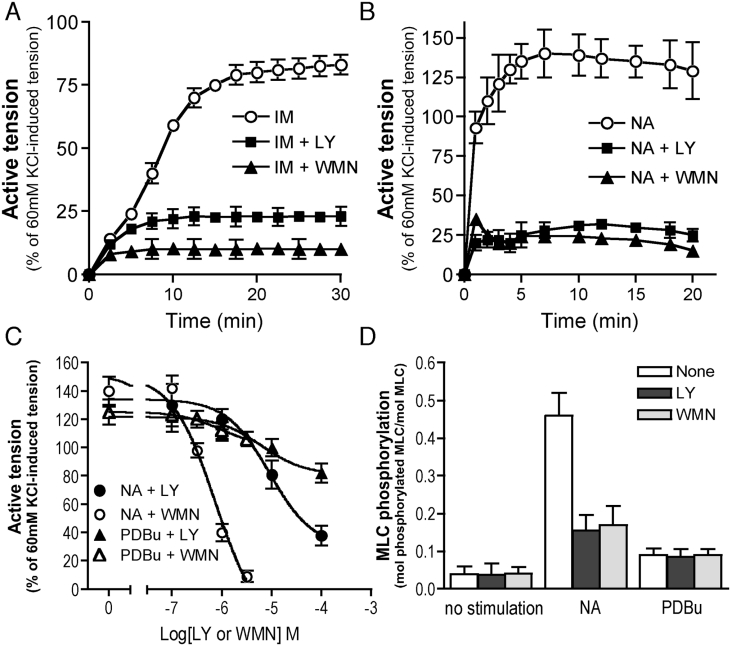
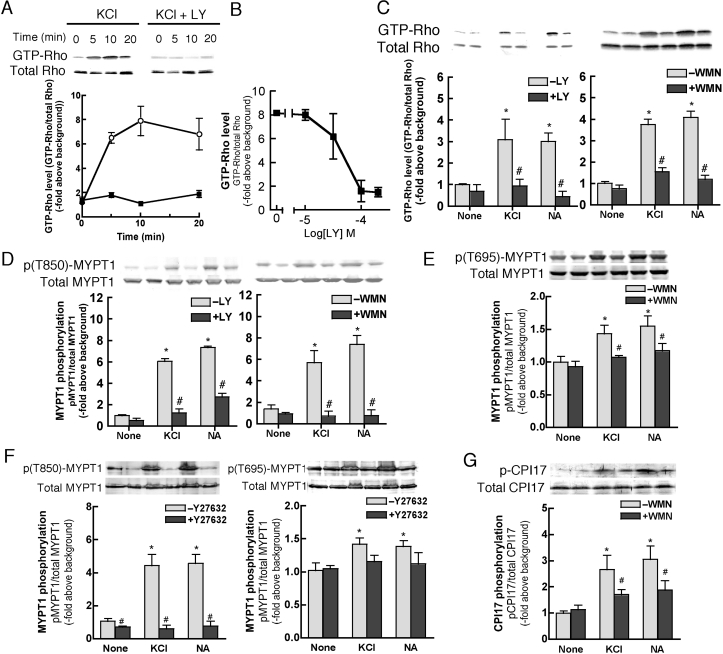
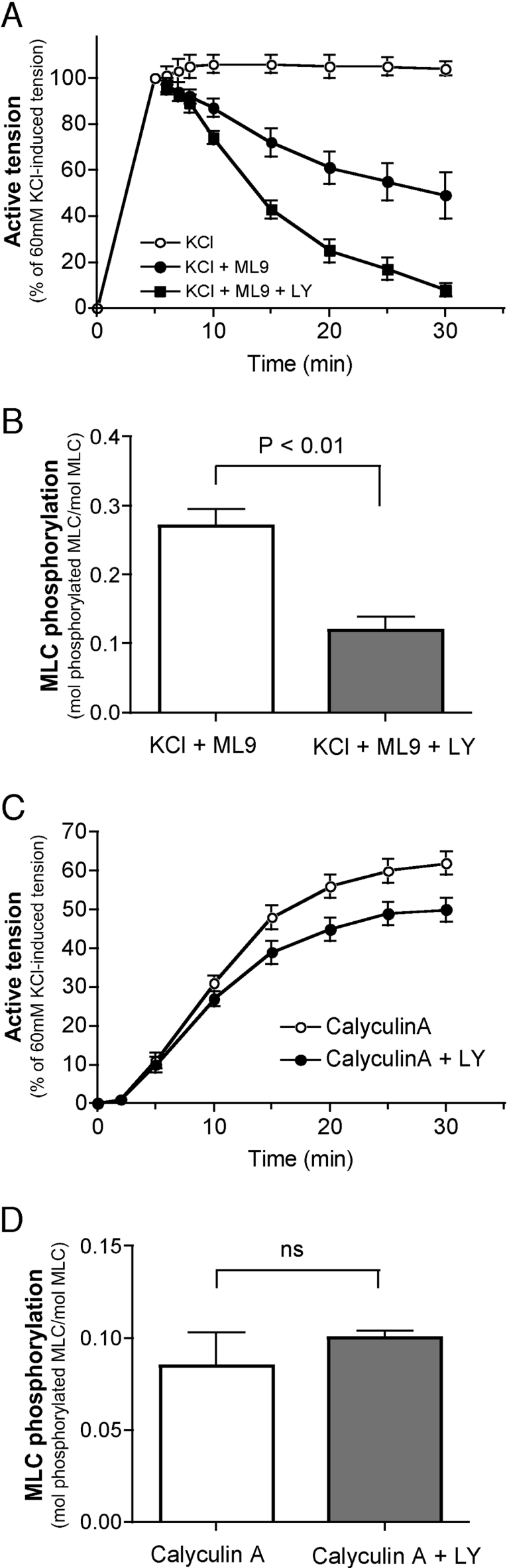
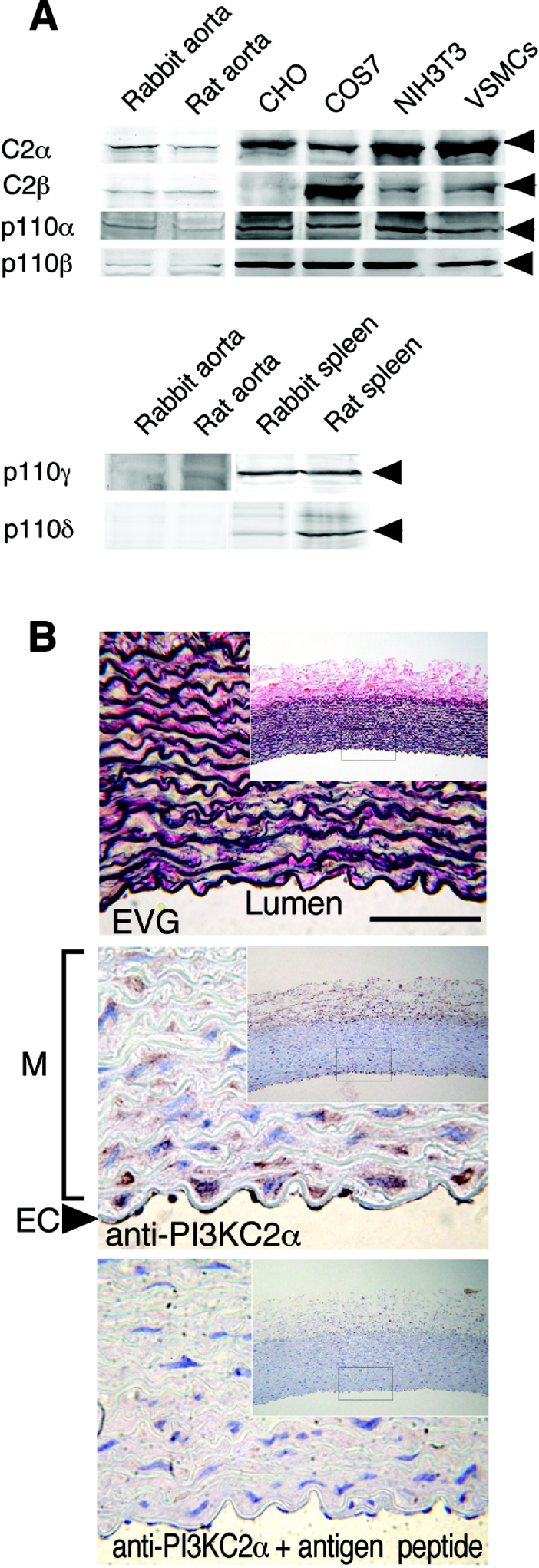
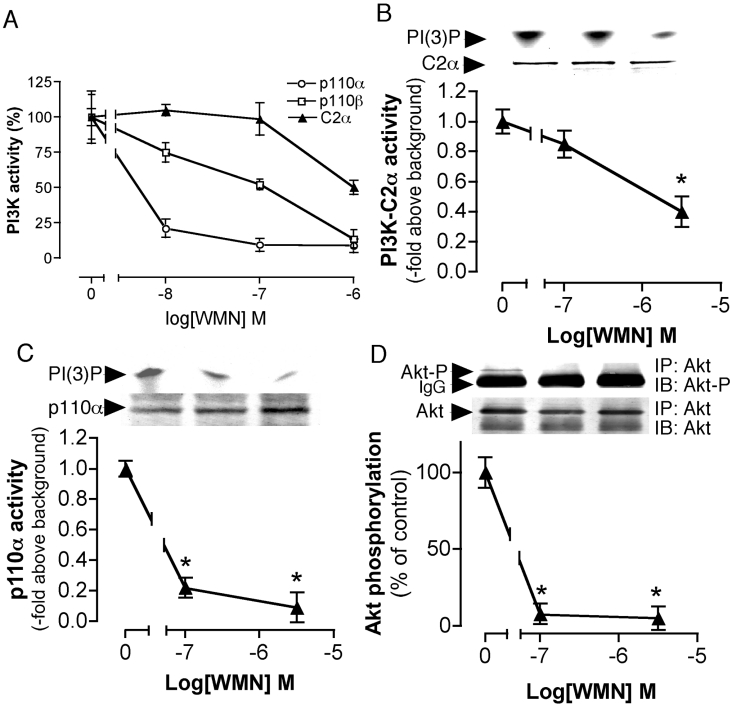
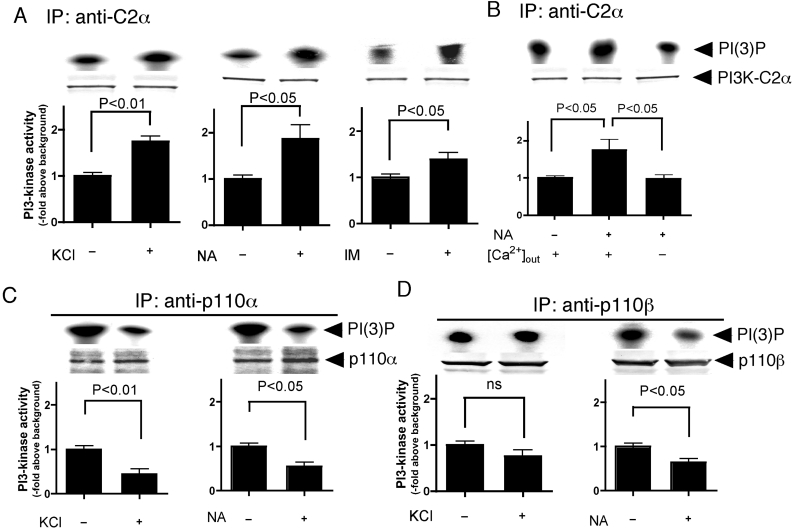
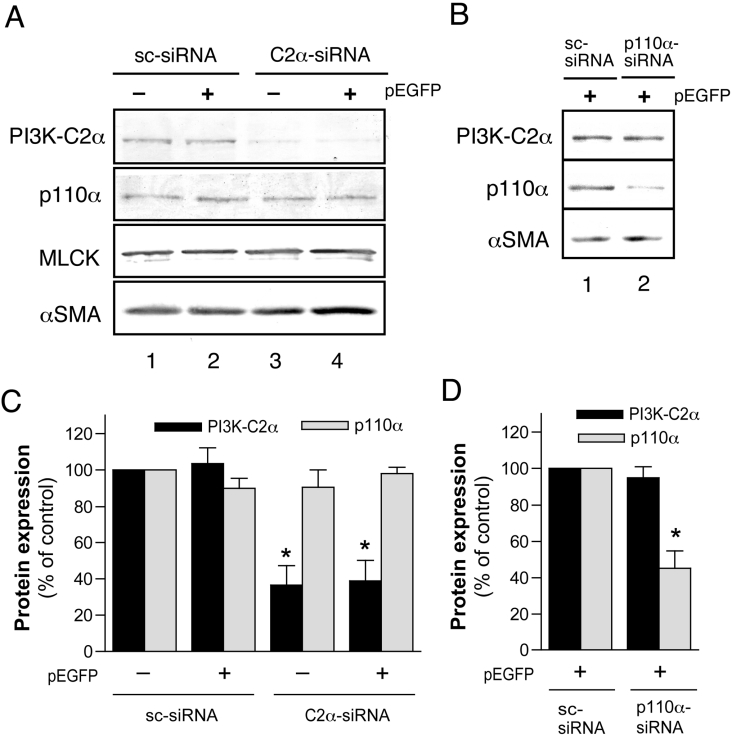
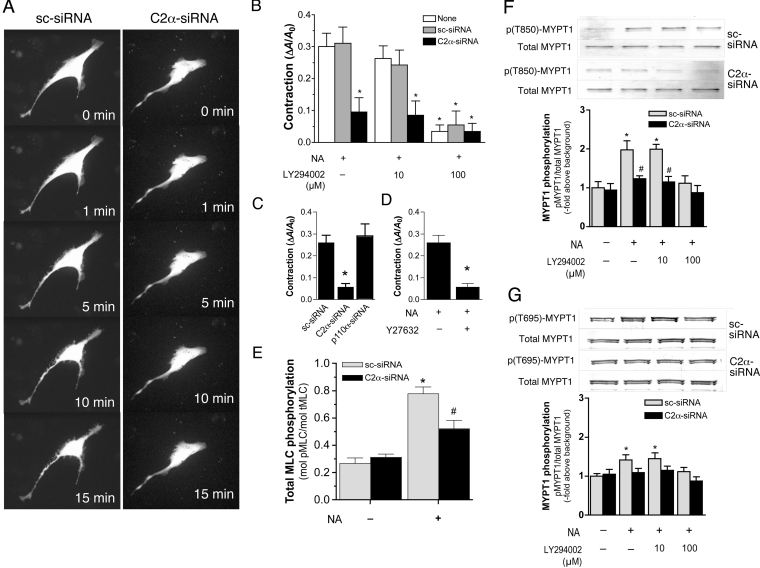
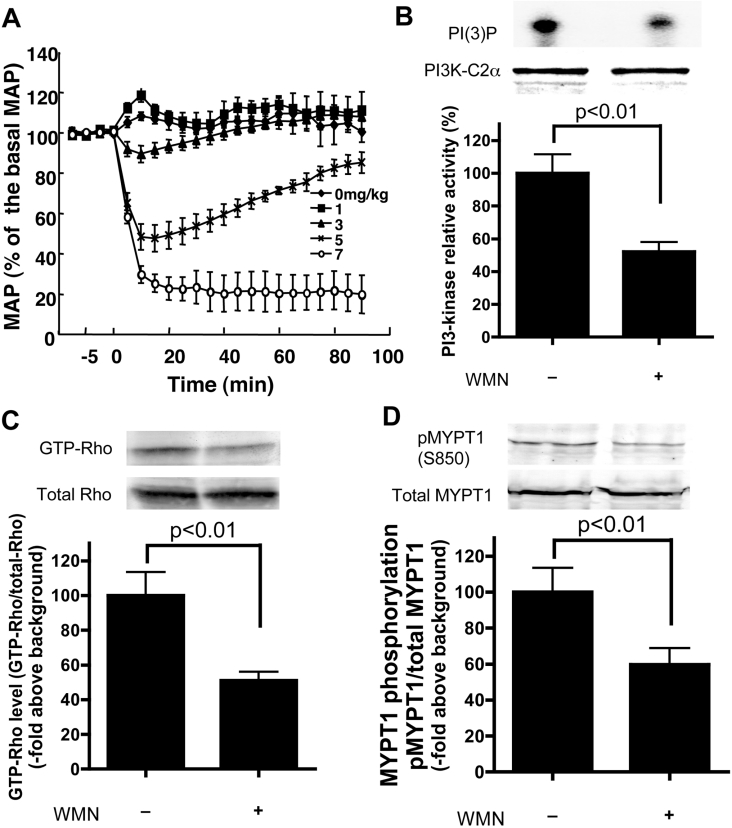
We demonstrated previously that membrane depolarization and excitatory receptor agonists such as noradrenaline induce Ca2+-dependent Rho activation in VSM (vascular smooth muscle), resulting in MP (myosin phosphatase) inhibition through the mechanisms involving Rho kinase-mediated phosphorylation of its regulatory subunit MYPT1. In the present study, we show in de-endothelialized VSM strips that the PI3K (phosphoinositide 3-kinase) inhibitors LY294002 and wortmannin inhibited KCl membrane depolarization- and noradrenaline-induced Rho activation and MYPT1 phosphorylation, with concomitant inhibition of MLC (20-kDa myosin light chain) phosphorylation and contraction. LY294002 also augmented de-phosphorylation of MLC and resultantly relaxation in KCl-contracted VSM, whereas LY294002 was much less effective or ineffective under the conditions in which MP was inhibited by either a phosphatase inhibitor or a phorbol ester in Rho-independent manners. VSM express at least four PI3K isoforms, including the class I enzymes p110alpha and p110beta and the class II enzymes PI3K-C2alpha and -C2beta. The dose-response relationships of PI3K-inhibitor-induced inhibition of Rho, MLC phosphorylation and contraction were similar to that of PI3K-C2alpha inhibition, but not to that of the class I PI3K inhibition. Moreover, KCl and noradrenaline induced stimulation of PI3K-C2alpha in a Ca2+-dependent manner, but not of p110alpha or p110beta. Down-regulation of PI3K-C2alpha expression by siRNA (small interfering RNA) inhibited contraction and phosphorylation of MYPT1 and MLC in VSM cells. Finally, intravenous wortmannin infusion induced sustained hypotension in rats, with inhibition of PI3K-C2alpha activity, GTP-loading of Rho and MYPT1 phosphorylation in the artery. These results indicate the novel role of PI3K-C2alpha in Ca2+-dependent Rho-mediated negative control of MP and thus VSM contraction.
Figures
References
-
- Somlyo A. P., Somlyo A. V. Signal transduction and regulation in smooth muscle. Nature (London) 1994;372:231–236. - PubMed
-
- Hartshorne D. J., Ito M., Ikebe M. Myosin and contractile activity in smooth muscle. Adv. Exp. Med. Biol. 1989;255:269–277. - PubMed
-
- Somlyo A. P., Somlyo A. V. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol. Rev. 2003;83:1325–1358. - PubMed
-
- Kitazawa T., Gaylinn B. D., Denney G. H., Somlyo A. P. G-protein-mediated Ca2+ sensitization of smooth muscle contraction through myosin light chain phosphorylation. J. Biol. Chem. 1991;266:1708–1715. - PubMed
-
- Noda M., Yasuda-Fukazawa C., Moriishi K., Kato T., Okuda T., Kurokawa K., Takuwa Y. Involvement of Rho in GTPγS-induced enhancement of phosphorylation of 20 kDa myosin light chain in vascular smooth muscle cells: inhibition of phosphatase activity. FEBS. Lett. 1995;367:246–250. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous