

Incomplete protein packing as a selectivity filter in drug design
- PMID: 16338411
- PMCID: PMC1963458
- DOI: 10.1016/j.str.2005.08.018
Incomplete protein packing as a selectivity filter in drug design
Abstract
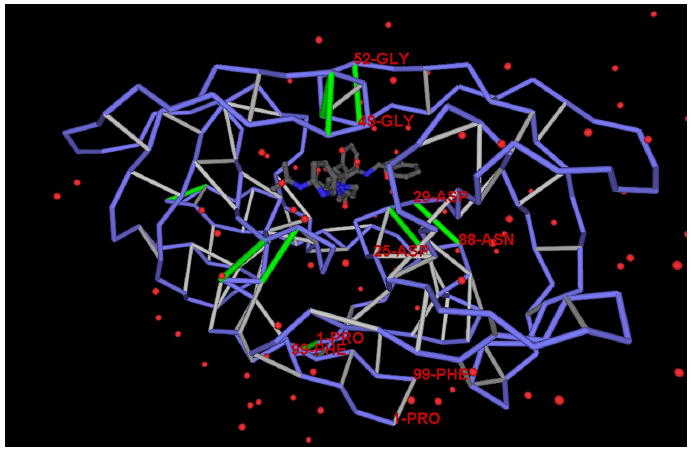
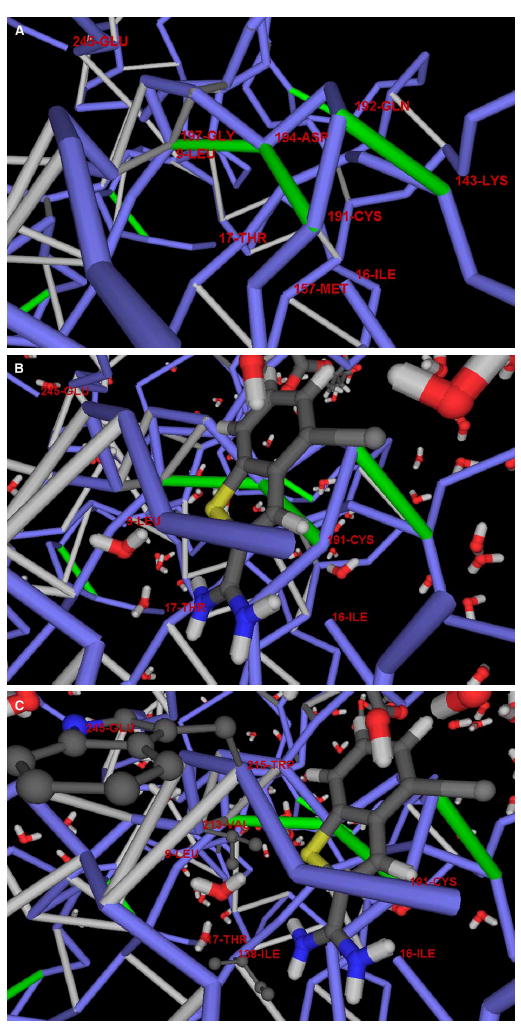
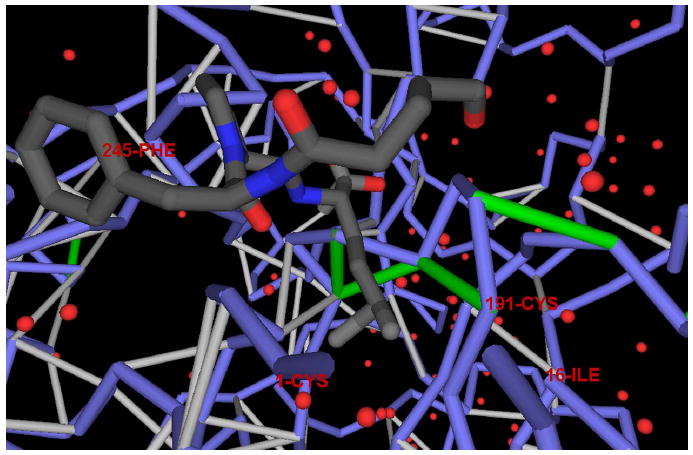
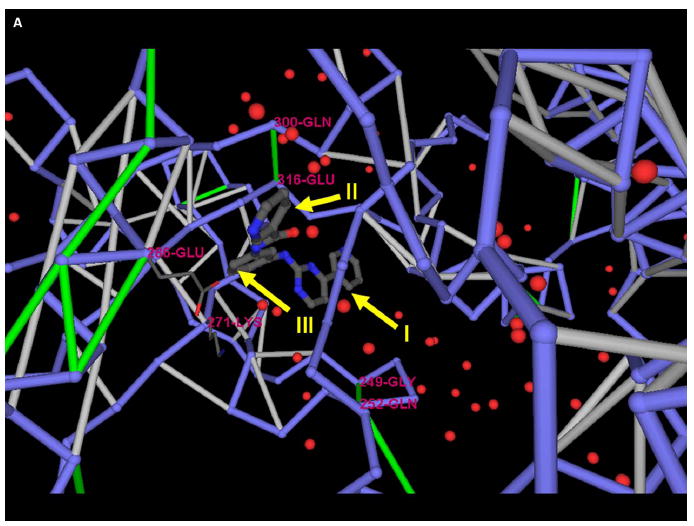
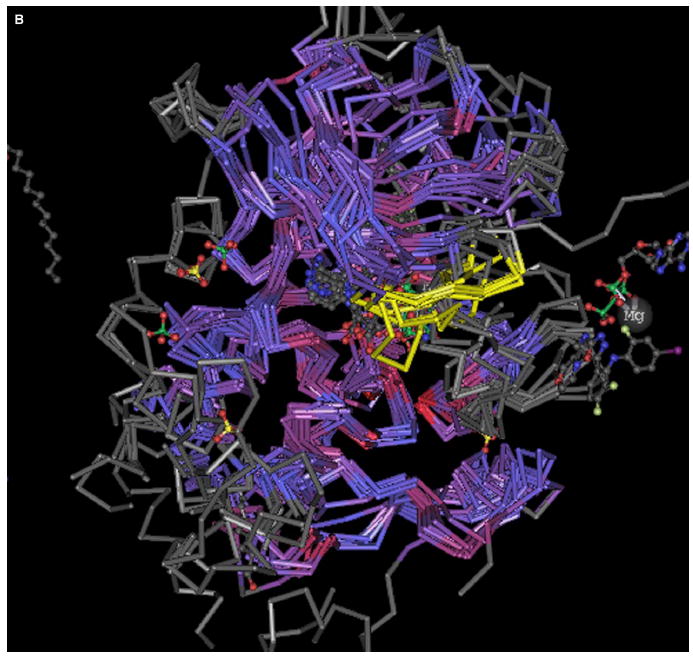
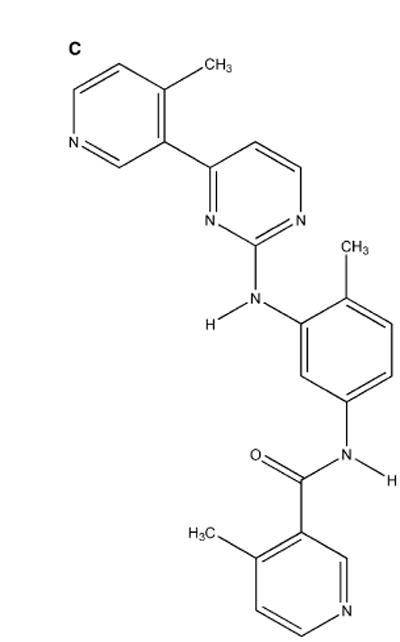
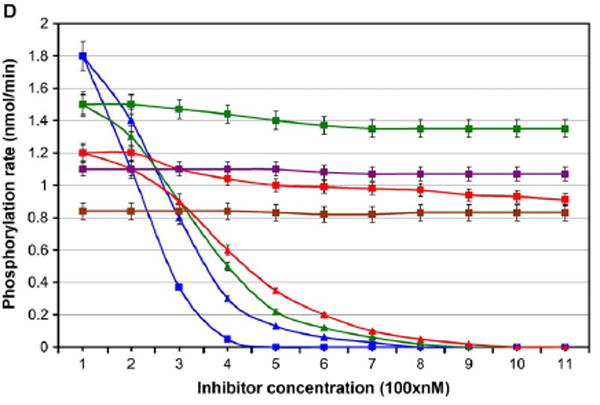
The conservation of structure across paralog proteins promotes alternative protein-ligand associations often leading to side effects in drug-based inhibition. However, sticky packing defects are typically not conserved across paralogs, making them suitable targets to reduce drug toxicity. This observation enables a strategy for the design of highly specific inhibitors involving ligands that wrap nonconserved packing defects. The selectivity of these inhibitors is evidenced in affinity assays on a cancer-related pharmacokinome: a powerful inhibitor is redesigned by using the wrapping technology to enhance its selectivity and affinity for a target kinase. In this way, the packing defects of a soluble protein may be used as selectivity filters for drug design.
Figures
Similar articles
-
Feature-similarity protein classifier as a ligand engineering tool.Biomol Eng. 2006 Dec;23(6):307-15. doi: 10.1016/j.bioeng.2006.09.004. Epub 2006 Oct 10. Biomol Eng. 2006. PMID: 17110166 Free PMC article.
-
Packing defects as selectivity switches for drug-based protein inhibitors.Proc Natl Acad Sci U S A. 2006 Jan 10;103(2):323-8. doi: 10.1073/pnas.0509351102. Epub 2005 Dec 30. Proc Natl Acad Sci U S A. 2006. Retraction in: Proc Natl Acad Sci U S A. 2006 Mar 14;103(11):4329. doi: 10.1073/pnas.0601034103. PMID: 16387853 Free PMC article. Retracted.
-
Taming the induced folding of drug-targeted kinases.Trends Pharmacol Sci. 2009 Feb;30(2):66-71. doi: 10.1016/j.tips.2008.11.001. Epub 2008 Dec 26. Trends Pharmacol Sci. 2009. PMID: 19097649
-
Wrapping technology and the enhancement of specificity in cancer drug treatment.Front Biosci. 2007 May 1;12:3617-27. doi: 10.2741/2338. Front Biosci. 2007. PMID: 17485325 Review.
-
Protein wrapping: a molecular marker for association, aggregation and drug design.Chem Soc Rev. 2008 Nov;37(11):2373-82. doi: 10.1039/b804150b. Epub 2008 Sep 15. Chem Soc Rev. 2008. PMID: 18949110 Review.
Cited by
-
Feature-similarity protein classifier as a ligand engineering tool.Biomol Eng. 2006 Dec;23(6):307-15. doi: 10.1016/j.bioeng.2006.09.004. Epub 2006 Oct 10. Biomol Eng. 2006. PMID: 17110166 Free PMC article.
-
Crystal structure of a bacterial phosphoglucomutase, an enzyme involved in the virulence of multiple human pathogens.Proteins. 2011 Apr;79(4):1215-29. doi: 10.1002/prot.22957. Epub 2011 Jan 18. Proteins. 2011. PMID: 21246636 Free PMC article.
-
Chemogenomic approaches to rational drug design.Br J Pharmacol. 2007 Sep;152(1):38-52. doi: 10.1038/sj.bjp.0707307. Epub 2007 May 29. Br J Pharmacol. 2007. PMID: 17533416 Free PMC article. Review.
-
Kinase packing defects as drug targets.Drug Discov Today. 2007 Nov;12(21-22):917-23. doi: 10.1016/j.drudis.2007.09.009. Epub 2007 Oct 30. Drug Discov Today. 2007. PMID: 17993409 Free PMC article. Review.
-
Inhibitor design for TMPRSS2: insights from computational analysis of its backbone hydrogen bonds using a simple descriptor.Eur Biophys J. 2024 Feb;53(1-2):27-46. doi: 10.1007/s00249-023-01695-4. Epub 2023 Dec 29. Eur Biophys J. 2024. PMID: 38157015 Free PMC article.
References
-
- Arkin MR, Wells JA. Small-molecule inhibitors of protein-protein interactions: progressing towards the dream. Nat Rev Drug Discov. 2004;3:301–317. - PubMed
-
- Attoub S, Rivat C, Rodrigues S, Van Borexlaer S, Bedin M, Buyneel E, Louvet C, Kornprobst M, Andre T, Mareel M, et al. The c-kit tyrosine kinase inhibitor STI-571 for colorectal cancer therapy. Cancer Res. 2002;62:4879–4883. - PubMed
-
- Deremble C, Lavery R. Macromolecular recognition. Curr Opin Struct Biol. 2005;15:171–175. - PubMed
-
- Dunker KA, Brown C, Obradovic Z. Indentification and function of usefully disordered proteins. Adv Protein Chem. 2002;62:25–49. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
