

Comparative genomic analyses of the bacterial phosphotransferase system
- PMID: 16339738
- PMCID: PMC1306802
- DOI: 10.1128/MMBR.69.4.608-634.2005
Comparative genomic analyses of the bacterial phosphotransferase system
Abstract
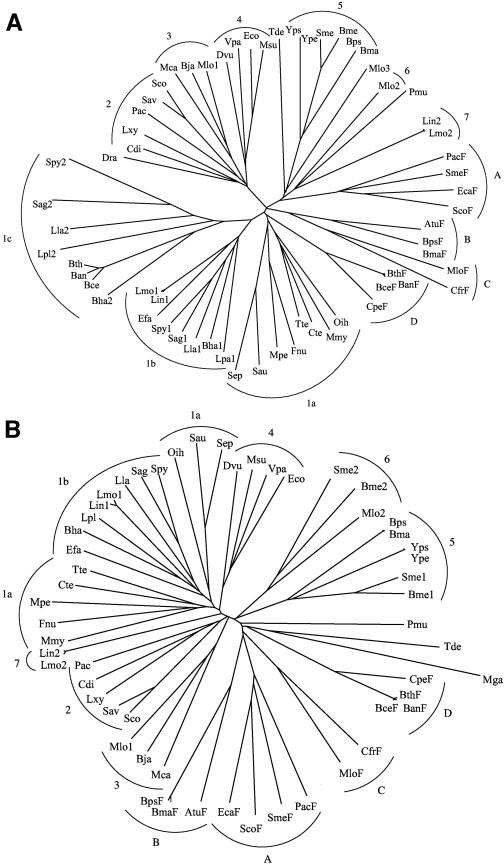
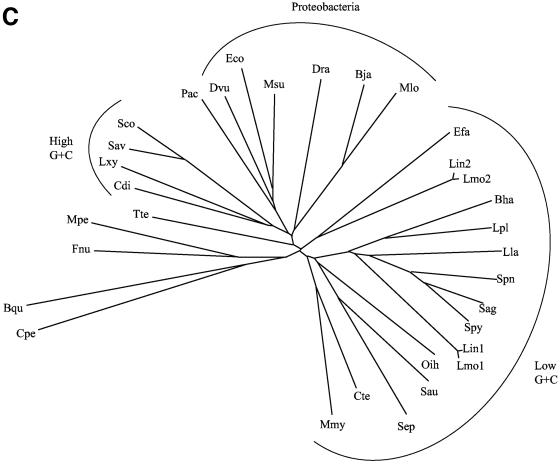
We report analyses of 202 fully sequenced genomes for homologues of known protein constituents of the bacterial phosphoenolpyruvate-dependent phosphotransferase system (PTS). These included 174 bacterial, 19 archaeal, and 9 eukaryotic genomes. Homologues of PTS proteins were not identified in archaea or eukaryotes, showing that the horizontal transfer of genes encoding PTS proteins has not occurred between the three domains of life. Of the 174 bacterial genomes (136 bacterial species) analyzed, 30 diverse species have no PTS homologues, and 29 species have cytoplasmic PTS phosphoryl transfer protein homologues but lack recognizable PTS permeases. These soluble homologues presumably function in regulation. The remaining 77 species possess all PTS proteins required for the transport and phosphorylation of at least one sugar via the PTS. Up to 3.2% of the genes in a bacterium encode PTS proteins. These homologues were analyzed for family association, range of protein types, domain organization, and organismal distribution. Different strains of a single bacterial species often possess strikingly different complements of PTS proteins. Types of PTS protein domain fusions were analyzed, showing that certain types of domain fusions are common, while others are rare or prohibited. Select PTS proteins were analyzed from different phylogenetic standpoints, showing that PTS protein phylogeny often differs from organismal phylogeny. The results document the frequent gain and loss of PTS protein-encoding genes and suggest that the lateral transfer of these genes within the bacterial domain has played an important role in bacterial evolution. Our studies provide insight into the development of complex multicomponent enzyme systems and lead to predictions regarding the types of protein-protein interactions that promote efficient PTS-mediated phosphoryl transfer.
Figures
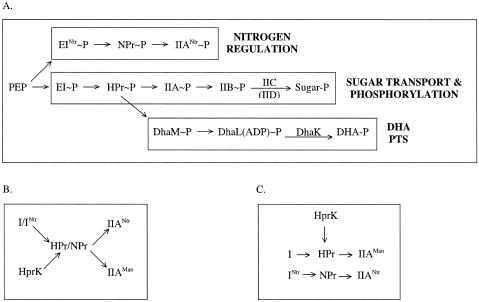
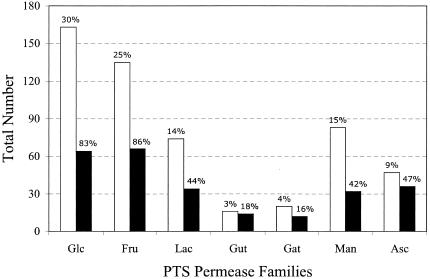
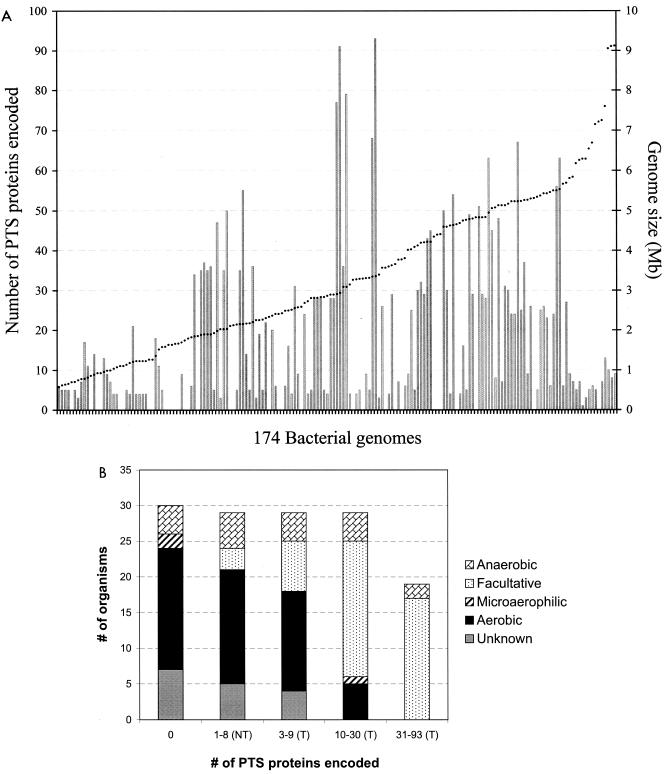
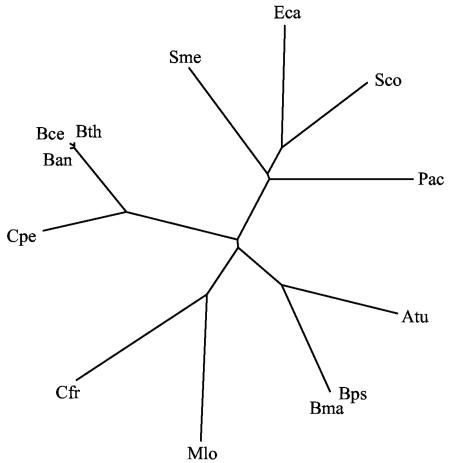
References
-
- Bächler, C. 2004. Catalysis and transcription control by the dihydroxyacetone kinase of Escherichia coli. Ph.D. thesis. University of Berne, Berne, Switzerland.
-
- Bächler, C., K. Flükiger-Brühwiler, P. Schneider, P. Bähler, and B. Erni. 2005. From ATP as substrate to ADP as coenzyme. Functional evolution of the nucleotide binding subunit of dihydroxyacetone kinases. J. Biol. Chem. 280:18321-18325. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
