

Regulation of Sar1 NH2 terminus by GTP binding and hydrolysis promotes membrane deformation to control COPII vesicle fission
- PMID: 16344311
- PMCID: PMC2171319
- DOI: 10.1083/jcb.200509095
Regulation of Sar1 NH2 terminus by GTP binding and hydrolysis promotes membrane deformation to control COPII vesicle fission
Abstract
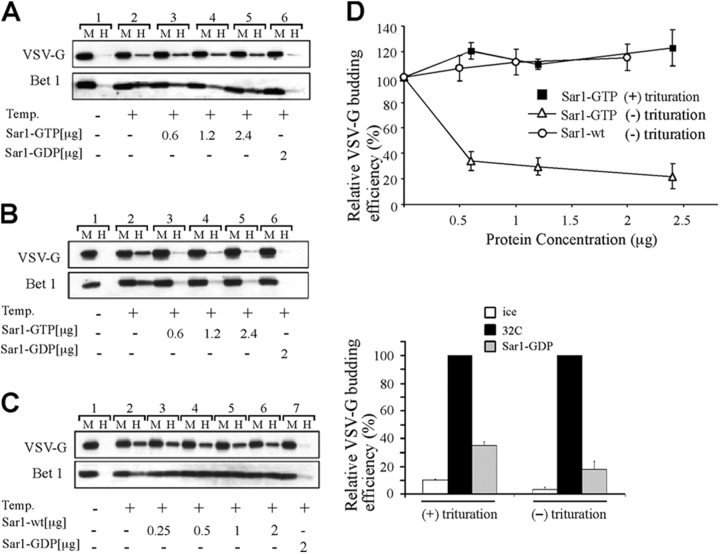
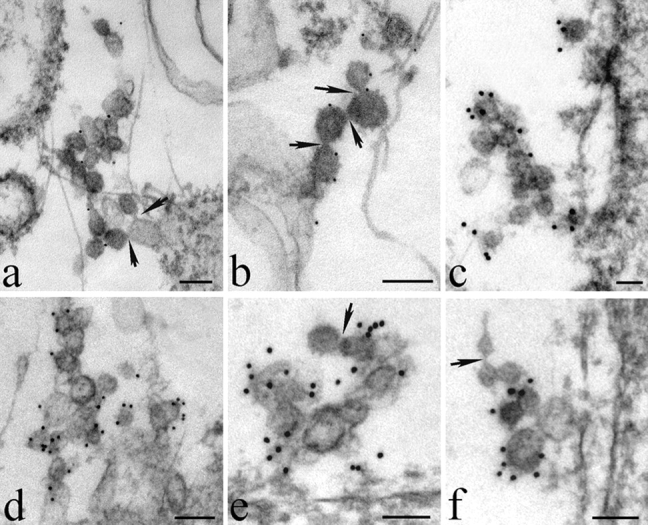
The mechanisms by which the coat complex II (COPII) coat mediates membrane deformation and vesicle fission are unknown. Sar1 is a structural component of the membrane-binding inner layer of COPII (Bi, X., R.A. Corpina, and J. Goldberg. 2002. Nature. 419:271-277). Using model liposomes we found that Sar1 uses GTP-regulated exposure of its NH2-terminal tail, an amphipathic peptide domain, to bind, deform, constrict, and destabilize membranes. Although Sar1 activation leads to constriction of endoplasmic reticulum (ER) membranes, progression to effective vesicle fission requires a functional Sar1 NH2 terminus and guanosine triphosphate (GTP) hydrolysis. Inhibition of Sar1 GTP hydrolysis, which stabilizes Sar1 membrane binding, resulted in the formation of coated COPII vesicles that fail to detach from the ER. Thus Sar1-mediated GTP binding and hydrolysis regulates the NH2-terminal tail to perturb membrane packing, promote membrane deformation, and control vesicle fission.
Figures


References
-
- Barlowe, C., L. Orci, T. Yeung, M. Hosobuchi, S. Hamamoto, N. Salama, M.F. Rexach, M. Ravazzola, M. Amherdt, and R. Schekman. 1994. COPII: a membrane coat formed by Sec proteins that drive vesicle budding from the endoplasmic reticulum. Cell. 77:895–907. - PubMed
