

NemaFootPrinter: a web based software for the identification of conserved non-coding genome sequence regions between C. elegans and C. briggsae
- PMID: 16351749
- PMCID: PMC1866385
- DOI: 10.1186/1471-2105-6-S4-S22
NemaFootPrinter: a web based software for the identification of conserved non-coding genome sequence regions between C. elegans and C. briggsae
Abstract
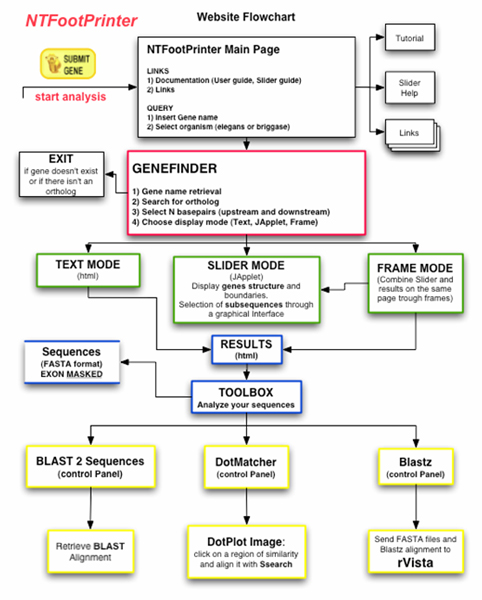
Background: NemaFootPrinter (Nematode Transcription Factor Scan Through Philogenetic Footprinting) is a web-based software for interactive identification of conserved, non-exonic DNA segments in the genomes of C. elegans and C. briggsae. It has been implemented according to the following project specifications:a) Automated identification of orthologous gene pairs. b) Interactive selection of the boundaries of the genes to be compared. c) Pairwise sequence comparison with a range of different methods. d) Identification of putative transcription factor binding sites on conserved, non-exonic DNA segments.
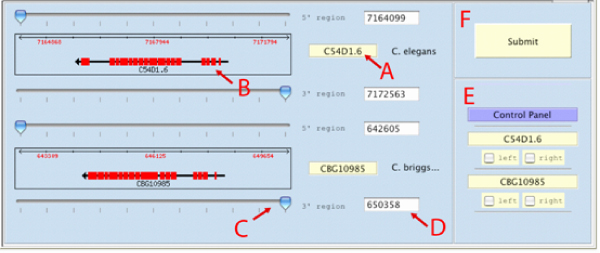
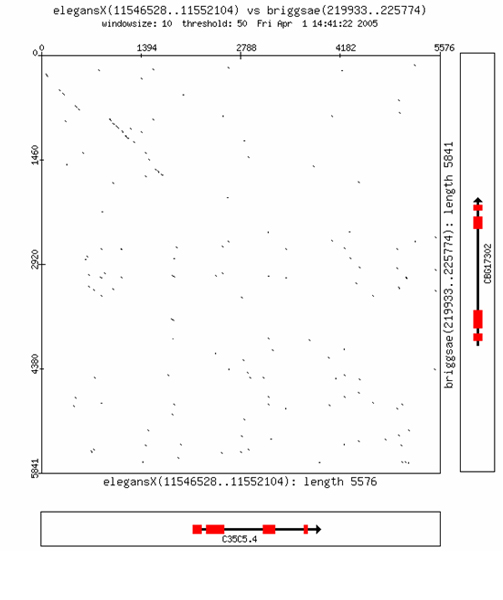
Results: Starting from a C. elegans or C. briggsae gene name or identifier, the software identifies the putative ortholog (if any), based on information derived from public nematode genome annotation databases. The investigator can then retrieve the genome DNA sequences of the two orthologous genes; visualize graphically the genes' intron/exon structure and the surrounding DNA regions; select, through an interactive graphical user interface, subsequences of the two gene regions. Using a bioinformatics toolbox (Blast2seq, Dotmatcher, Ssearch and connection to the rVista database) the investigator is able at the end of the procedure to identify and analyze significant sequences similarities, detecting the presence of transcription factor binding sites corresponding to the conserved segments. The software automatically masks exons.
Discussion: This software is intended as a practical and intuitive tool for the researchers interested in the identification of non-exonic conserved sequence segments between C. elegans and C. briggsae. These sequences may contain regulatory transcriptional elements since they are conserved between two related, but rapidly evolving genomes. This software also highlights the power of genome annotation databases when they are conceived as an open resource and the possibilities offered by seamless integration of different web services via the http protocol.
Availability: The program is freely available at http://bio.ifom-firc.it/NTFootPrinter.
Figures
Similar articles
-
CisOrtho: a program pipeline for genome-wide identification of transcription factor target genes using phylogenetic footprinting.BMC Bioinformatics. 2004 Mar 12;5:27. doi: 10.1186/1471-2105-5-27. BMC Bioinformatics. 2004. PMID: 15113408 Free PMC article.
-
The genome sequence of Caenorhabditis briggsae: a platform for comparative genomics.PLoS Biol. 2003 Nov;1(2):E45. doi: 10.1371/journal.pbio.0000045. Epub 2003 Nov 17. PLoS Biol. 2003. PMID: 14624247 Free PMC article.
-
Large synteny blocks revealed between Caenorhabditis elegans and Caenorhabditis briggsae genomes using OrthoCluster.BMC Genomics. 2010 Sep 24;11:516. doi: 10.1186/1471-2164-11-516. BMC Genomics. 2010. PMID: 20868500 Free PMC article.
-
Comparative genomics in C. elegans, C. briggsae, and other Caenorhabditis species.Methods Mol Biol. 2006;351:13-29. doi: 10.1385/1-59745-151-7:13. Methods Mol Biol. 2006. PMID: 16988423 Review.
-
The draft genome sequence of the nematode Caenorhabditis briggsae, a companion to C. elegans.Genome Biol. 2003;4(12):238. doi: 10.1186/gb-2003-4-12-238. Epub 2003 Nov 18. Genome Biol. 2003. PMID: 14659008 Free PMC article. Review.
Cited by
-
Phylogenetic simulation of promoter evolution: estimation and modeling of binding site turnover events and assessment of their impact on alignment tools.Genome Biol. 2007;8(10):R225. doi: 10.1186/gb-2007-8-10-r225. Genome Biol. 2007. PMID: 17956628 Free PMC article.
References
-
- Boccia A, Petrillo M, di Bernardo D, Guffanti A, Mignone F, Confalonieri S, Luzi L, Pesole G, Paolella G, Ballabio A, Banfi S. DG-CST (Disease Gene Conserved Sequence Tags), a database of human-mouse conserved elements associated to disease genes. Nucleic Acids Res. 2005;33:D505–510. doi: 10.1093/nar/gki011. - DOI - PMC - PubMed
