

Repression of beta-catenin function in malignant cells by nonsteroidal antiinflammatory drugs
- PMID: 16352713
- PMCID: PMC1317972
- DOI: 10.1073/pnas.0509316102
Repression of beta-catenin function in malignant cells by nonsteroidal antiinflammatory drugs
Abstract
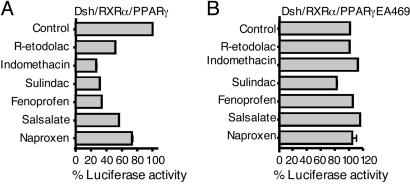
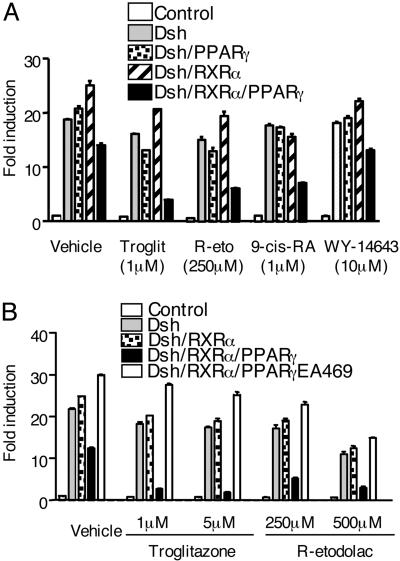
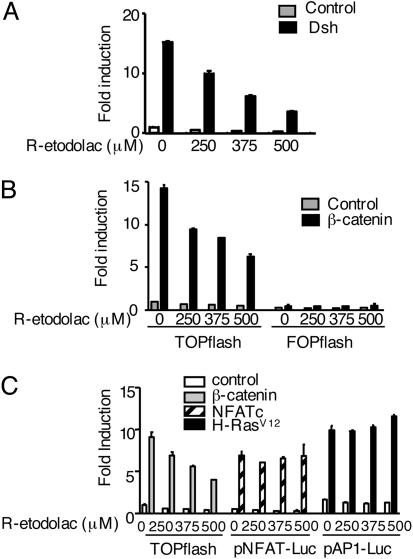
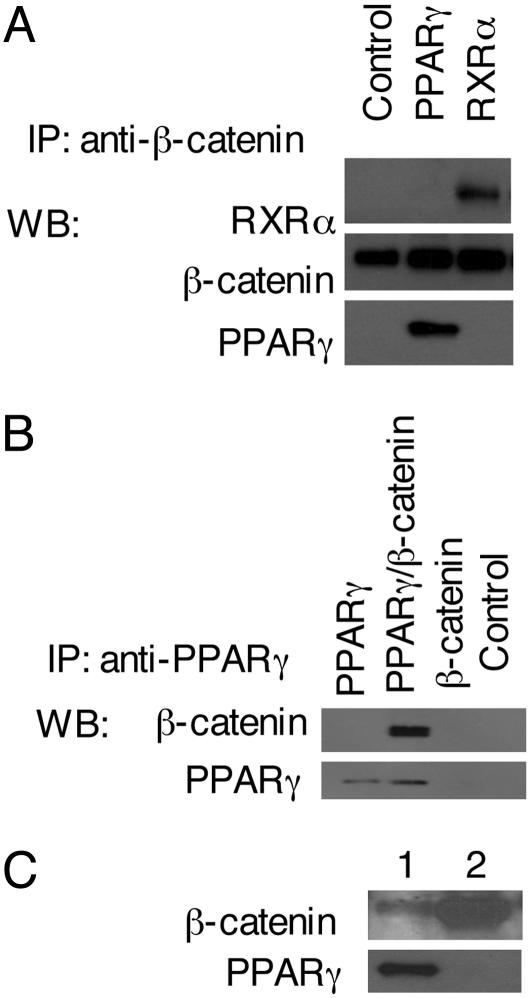
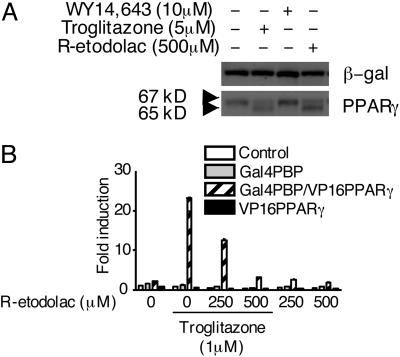
Activation of the Wnt/beta-catenin pathway promotes the development of several cancers and is an attractive target for chemopreventive and chemotherapeutic agents. Nonsteroidal antiinflammatory drugs (NSAIDs) have been reported to antagonize beta-catenin function, but their mechanism of action is not known. We demonstrate here that interference with beta-catenin function by NSAIDs does not correlate with cyclooxygenase (COX) inhibition. Instead, NSAID inhibition of beta-catenin requires the high level expression of peroxisome proliferator-activated receptor gamma (PPAR-gamma) and its co-receptor retinoid-X-receptor alpha (RXR-alpha). Immunoprecipitation experiments show that beta-catenin interacts with RXR-alpha and PPAR-gamma in some malignant cells. Repression of beta-catenin-dependent transcription by NSAIDs is thus indirect and depends on the coexpression of other nuclear receptors.
Figures
Similar articles
-
Regulation of peroxisome proliferator-activated receptor-beta/delta by the APC/beta-CATENIN pathway and nonsteroidal antiinflammatory drugs.Mol Carcinog. 2009 Oct;48(10):942-52. doi: 10.1002/mc.20546. Mol Carcinog. 2009. PMID: 19415698 Free PMC article.
-
The nonsteroidal anti-inflammatory drugs aspirin and indomethacin attenuate beta-catenin/TCF-4 signaling.Oncogene. 2001 Feb 1;20(5):645-53. doi: 10.1038/sj.onc.1204123. Oncogene. 2001. PMID: 11313997
-
R-Etodolac decreases beta-catenin levels along with survival and proliferation of hepatoma cells.J Hepatol. 2007 May;46(5):849-57. doi: 10.1016/j.jhep.2006.11.017. Epub 2006 Dec 21. J Hepatol. 2007. PMID: 17275129 Free PMC article.
-
Targeting the Canonical WNT/β-Catenin Pathway in Cancer Treatment Using Non-Steroidal Anti-Inflammatory Drugs.Cells. 2019 Jul 15;8(7):726. doi: 10.3390/cells8070726. Cells. 2019. PMID: 31311204 Free PMC article. Review.
-
Interaction of nuclear receptors with the Wnt/beta-catenin/Tcf signaling axis: Wnt you like to know?Endocr Rev. 2005 Dec;26(7):898-915. doi: 10.1210/er.2003-0034. Epub 2005 Aug 26. Endocr Rev. 2005. PMID: 16126938 Review.
Cited by
-
Molecular Mechanisms of the Antitumor Effects of Mesalazine and Its Preventive Potential in Colorectal Cancer.Molecules. 2023 Jun 29;28(13):5081. doi: 10.3390/molecules28135081. Molecules. 2023. PMID: 37446747 Free PMC article. Review.
-
Synergistic Effects of PPARgamma Ligands and Retinoids in Cancer Treatment.PPAR Res. 2008;2008:181047. doi: 10.1155/2008/181047. PPAR Res. 2008. PMID: 18528526 Free PMC article.
-
Spiperone enhances intracellular calcium level and inhibits the Wnt signaling pathway.BMC Pharmacol. 2009 Nov 30;9:13. doi: 10.1186/1471-2210-9-13. BMC Pharmacol. 2009. PMID: 19948059 Free PMC article.
-
Distinct patterns of ALDH1A1 expression predict metastasis and poor outcome of colorectal carcinoma.Int J Clin Exp Pathol. 2014 May 15;7(6):2976-86. eCollection 2014. Int J Clin Exp Pathol. 2014. PMID: 25031716 Free PMC article.
-
Regression of sporadic intra-abdominal desmoid tumour following administration of non-steroidal anti-inflammatory drug.World J Surg Oncol. 2008 Feb 8;6:17. doi: 10.1186/1477-7819-6-17. World J Surg Oncol. 2008. PMID: 18257933 Free PMC article.
References
-
- Harris, R. E., Chlebowski, R. T., Jackson, R. D., Frid, D. J., Ascenseo, J. L., Anderson, G., Loar, A., Rodabough, R. J., White, E. & McTiernan, A. (2003) Cancer Res. 63, 6096-6101. - PubMed
-
- Irani, J., Ravery, V., Pariente, J. L., Chartier-Kastler, E., Lechevallier, E., Soulie, M., Chautard, D., Coloby, P., Fontaine, E., Bladou, F., et al. (2002) J. Urol. 168, 1985-1988. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
