

Differentiation of naive cord-blood T cells into CD19-specific cytolytic effectors for posttransplantation adoptive immunotherapy
- PMID: 16352804
- PMCID: PMC1895371
- DOI: 10.1182/blood-2005-09-3904
Differentiation of naive cord-blood T cells into CD19-specific cytolytic effectors for posttransplantation adoptive immunotherapy
Abstract
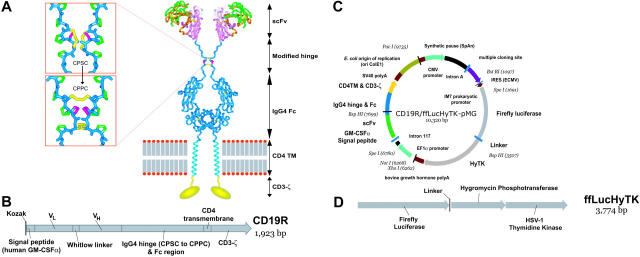
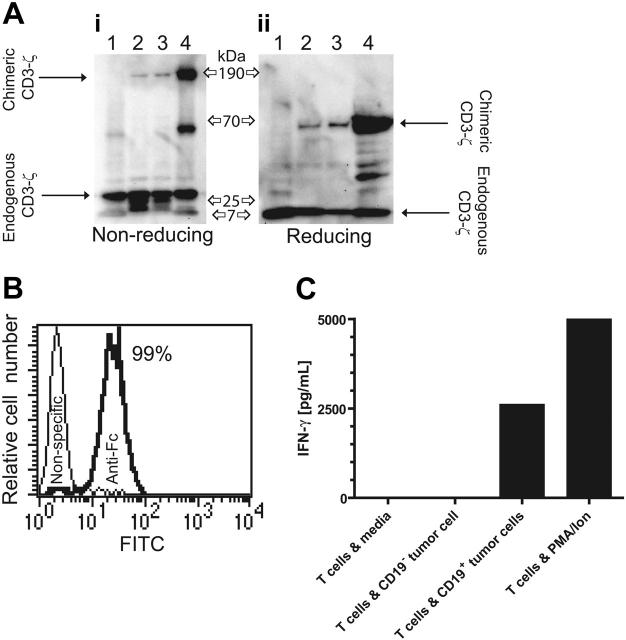
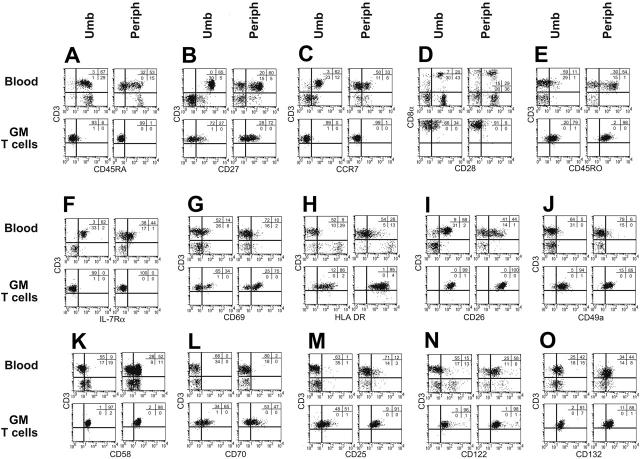
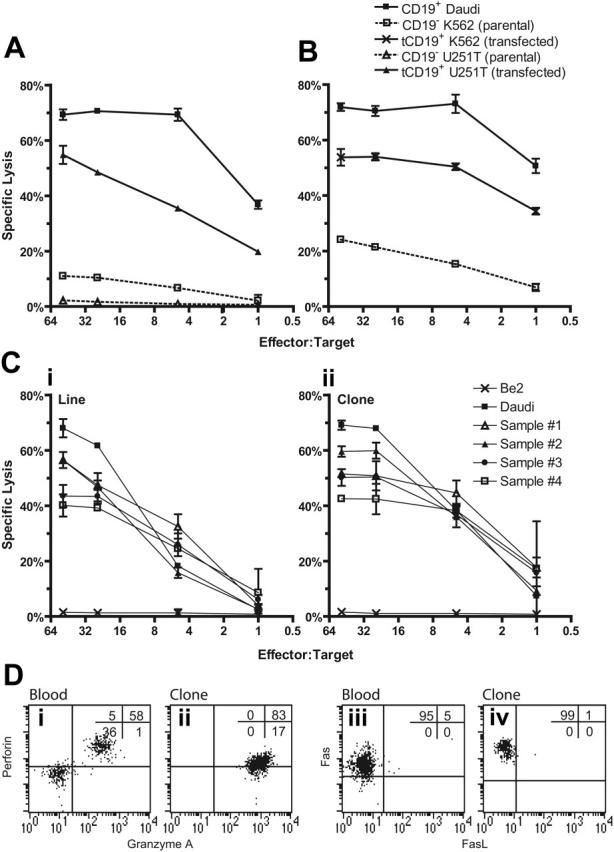
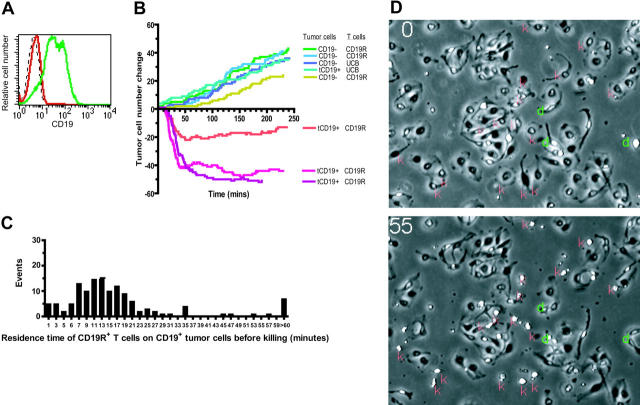
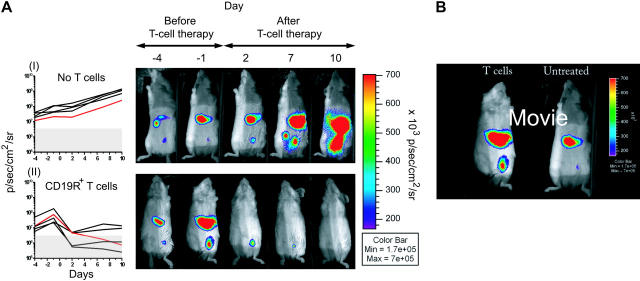
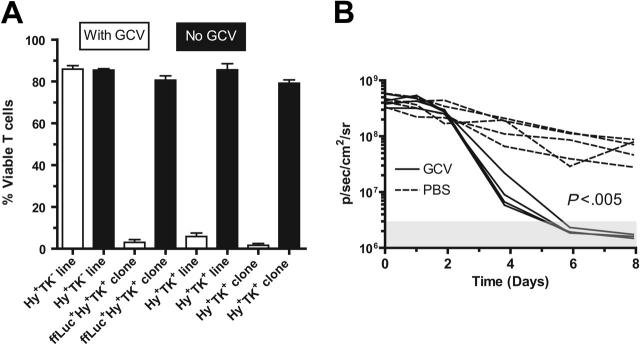
Disease relapse is a barrier to achieving therapeutic success after unrelated umbilical cord-blood transplantation (UCBT) for B-lineage acute lymphoblastic leukemia (B-ALL). While adoptive transfer of donor-derived tumor-specific T cells is a conceptually attractive approach to eliminating residual disease after allogeneic hematopoietic stem cell transplantation, adoptive immunotherapy after UCBT is constrained by the difficulty of generating antigen-specific T cells from functionally naive umbilical cord-blood (UCB)-derived T cells. Therefore, to generate T cells that recognize B-ALL, we have developed a chimeric immunoreceptor to redirect the specificity of T cells for CD19, a B-lineage antigen, and expressed this transgene in UCB-derived T cells. An ex vivo process, which is compliant with current good manufacturing practice for T-cell trials, has been developed to genetically modify and numerically expand UCB-derived T cells into CD19-specific effector cells. These are capable of CD19-restricted cytokine production and cytolysis in vitro, as well as mediating regression of CD19+ tumor and being selectively eliminated in vivo. Moreover, time-lapse microscopy of the genetically modified T-cell clones revealed an ability to lyse CD19+ tumor cells specifically and repetitively. These data provide the rationale for infusing UCB-derived CD19-specific T cells after UCBT to reduce the incidence of CD19+ B-ALL relapse.
Figures
References
-
- Rubinstein P, Carrier C, Scaradavou A, et al. Outcomes among 562 recipients of placental-blood transplants from unrelated donors. N Engl J Med. 1998;339: 1565-1577. - PubMed
-
- Porter DL, June CH. T-cell reconstitution and expansion after hematopoietic stem cell transplantation:'T' it up! Bone Marrow Transplant. 2005;35: 935-942. - PubMed
-
- Warren EH, Gavin M, Greenberg PD, Riddell SR. Minor histocompatibility antigens as targets for T-cell therapy after bone marrow transplantation. Curr Opin Hematol. 1998;5: 429-433. - PubMed
-
- Nijmeijer BA, Willemze R, Falkenburg JH. An animal model for human cellular immunotherapy: specific eradication of human acute lymphoblastic leukemia by cytotoxic T lymphocytes in NOD/scid mice. Blood. 2002;100: 654-660. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
