

Role of poly(ADP-ribose) polymerase-1 activation in the pathogenesis of diabetic complications: endothelial dysfunction, as a common underlying theme
- PMID: 16356120
- PMCID: PMC2228261
- DOI: 10.1089/ars.2005.7.1568
Role of poly(ADP-ribose) polymerase-1 activation in the pathogenesis of diabetic complications: endothelial dysfunction, as a common underlying theme
Abstract
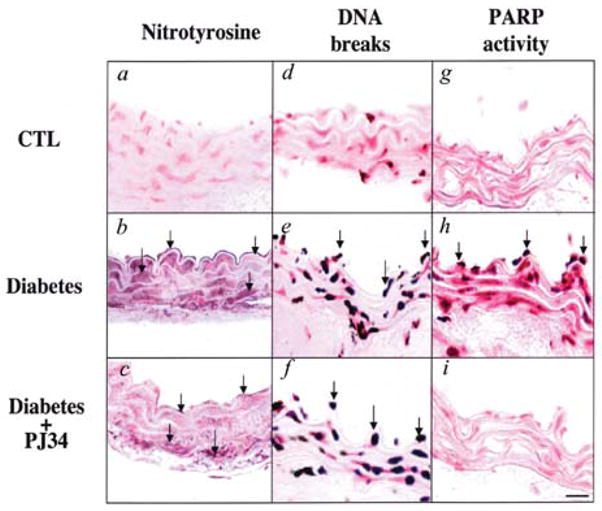
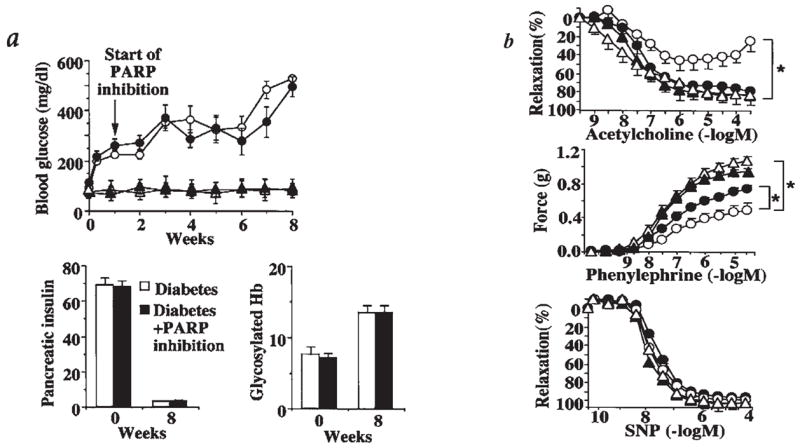
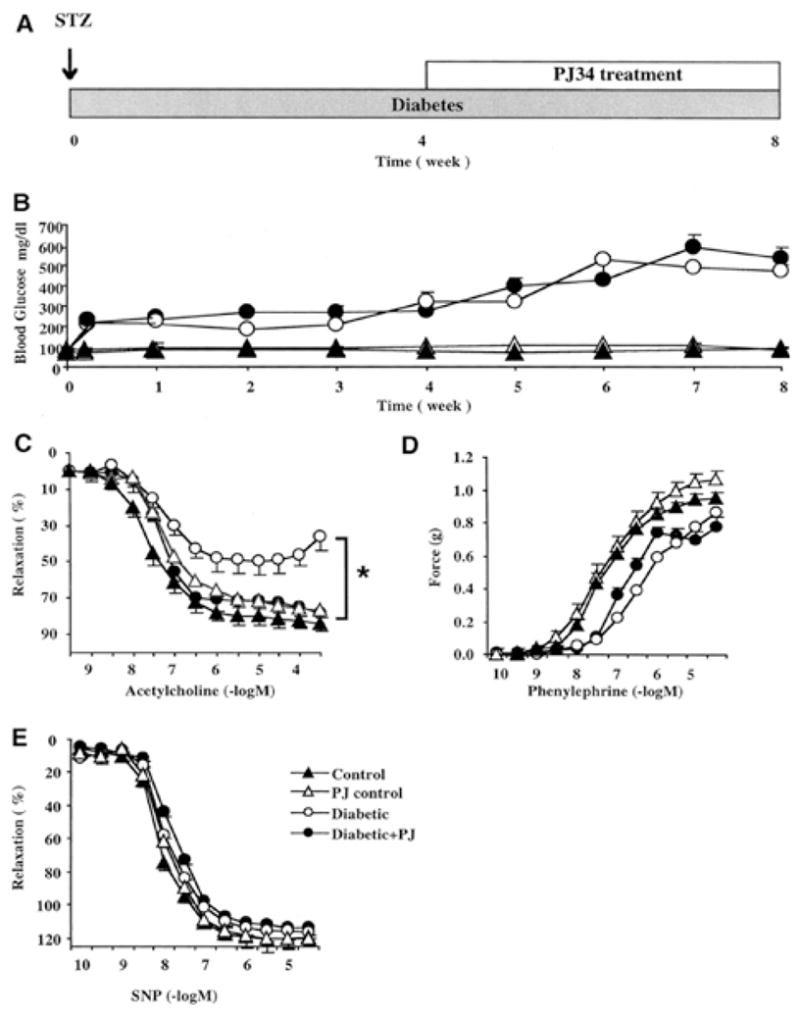
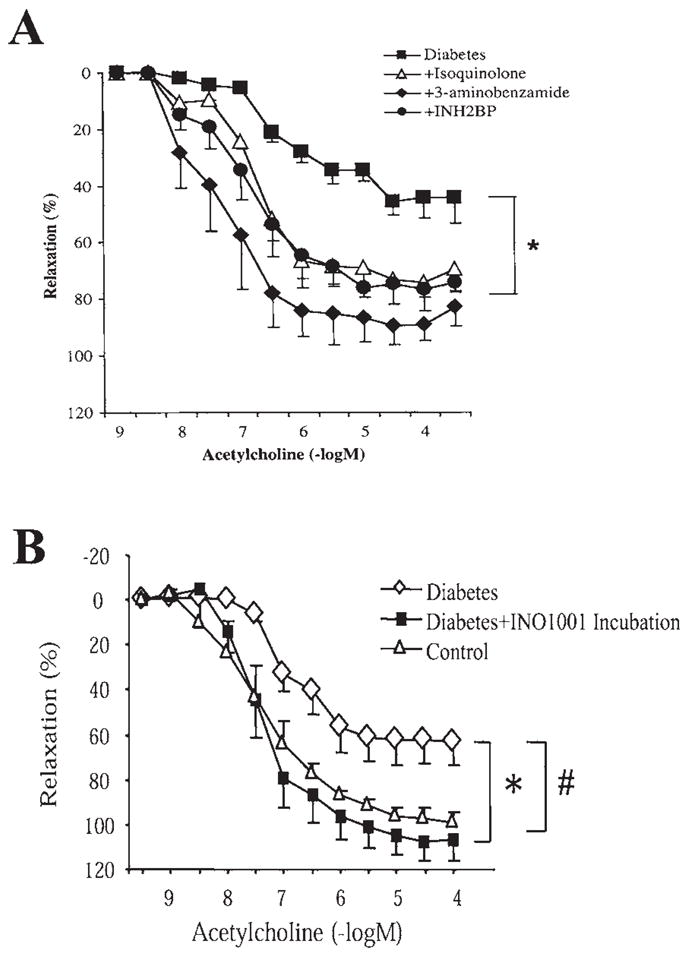
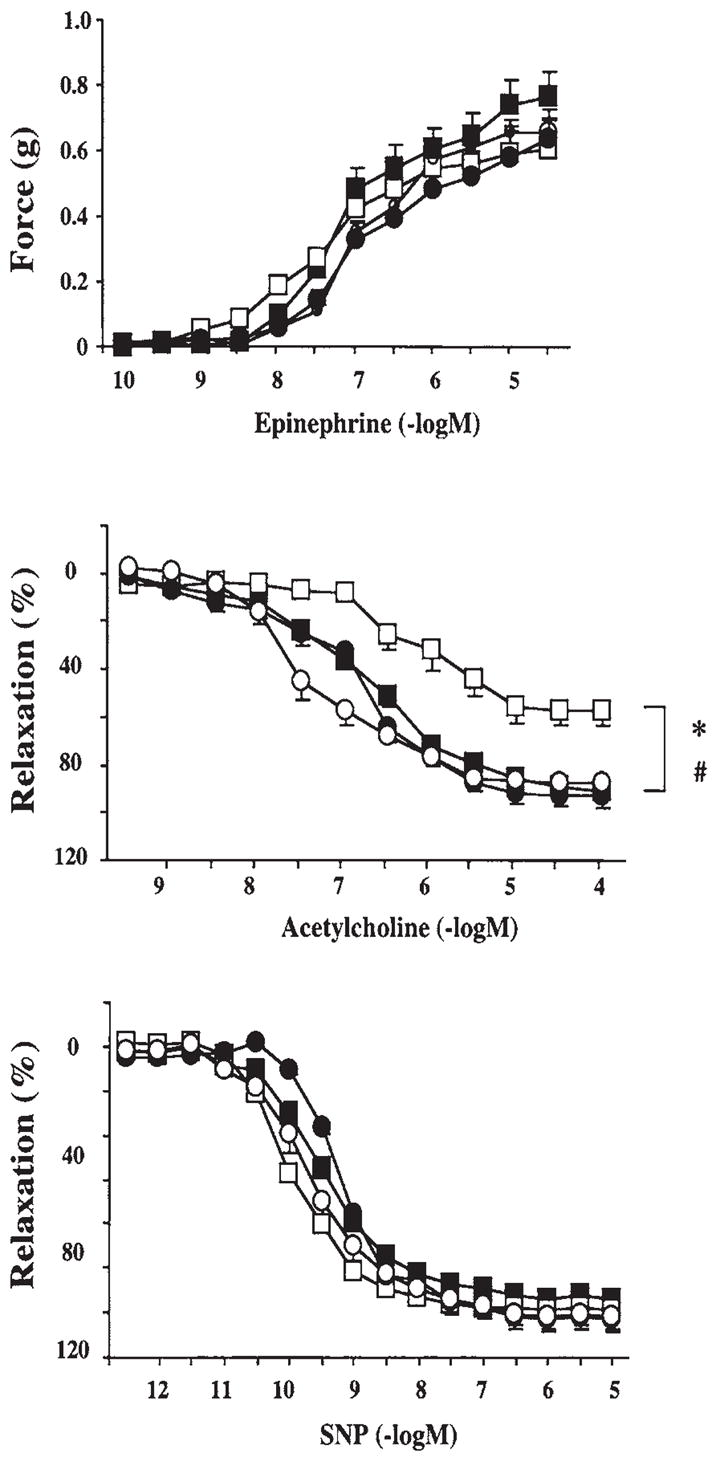
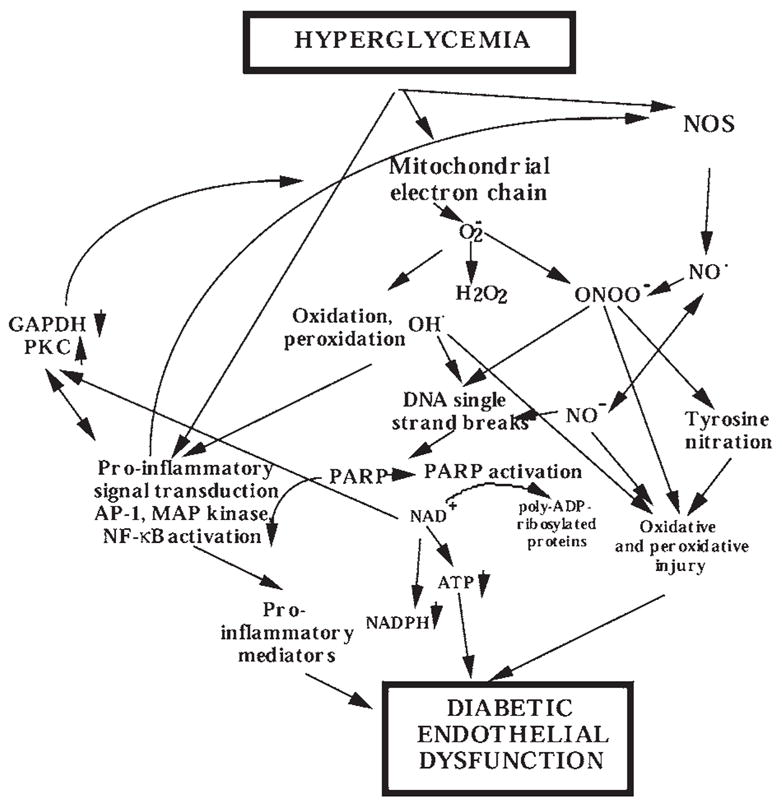
Hyperglycemia-induced overproduction of superoxide by mitochondrial electron-transport chain triggers several pathways of injury involved in the pathogenesis of diabetic complications [protein kinase C (PKC), hexosamine and polyol pathway fluxes, advanced glycation end product (AGE) formation] by inhibiting glyceraldehyde- 3-phosphate dehydrogenase (GAPDH) activity. Increased oxidative and nitrosative stress activates the nuclear enzyme, poly(ADP-ribose) polymerase-1 (PARP). PARP activation, on the one hand, depletes its substrate, NAD+, slowing the rate of glycolysis, electron transport, and ATP formation. On the other hand, it inhibits GAPDH by poly(ADP-ribosy)lation. These processes result in acute endothelial dysfunction in diabetic blood vessels, which importantly contributes to the development of various diabetic complications. Accordingly, hyperglycemia-induced activation of PKC isoforms, hexosaminase pathway flux, and AGE formation is prevented by blocking PARP activity. Furthermore, inhibition of PARP protects against diabetic cardiovascular dysfunction in preclinical models. PARP activation is present in microvasculature of human diabetic subjects. The oxidative/nitrosative stress-PARP pathway leads to diabetes-induced endothelial dysfunction, which may be an important underlying mechanism for the pathogenesis of other diabetic complications (cardiomyopathy, nephropathy, neuropathy, and retinopathy). This review focuses on the role of PARP in diabetic complications and the unique therapeutic potential of PARP inhibition in the prevention or reversal of diabetic complications.
Antioxid. Redox Signal. 7, 1568-1580.
Figures
References
-
- Anderson D, Yu TW, Wright J, Ioannides C. An examination of DNA strand breakage in the comet assay and antioxidant capacity in diabetic patients. Mutat Res. 1998;398:151–161. - PubMed
-
- Andreoli SP. Mechanisms of endothelial cell ATP depletion after oxidant injury. Pediatr Res. 1989;25:97–101. - PubMed
-
- Astley S, Langrish-Smith A, Southon S, Sampson M. Vitamin E supplementation and oxidative damage to DNA and plasma LDL in type 1 diabetes. Diabetes Care. 1999;22:1626–1631. - PubMed
-
- Beckman JA. Inhibition of protein kinase Cbeta prevents impaired endothelium-dependent vasodilation caused by hyperglycemia in humans. Circ Res. 2002;90:107–111. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
