

SLC34A3 mutations in patients with hereditary hypophosphatemic rickets with hypercalciuria predict a key role for the sodium-phosphate cotransporter NaPi-IIc in maintaining phosphate homeostasis
- PMID: 16358214
- PMCID: PMC1380228
- DOI: 10.1086/499409
SLC34A3 mutations in patients with hereditary hypophosphatemic rickets with hypercalciuria predict a key role for the sodium-phosphate cotransporter NaPi-IIc in maintaining phosphate homeostasis
Abstract
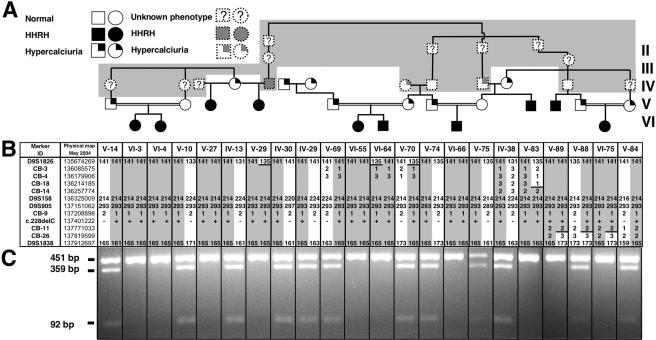
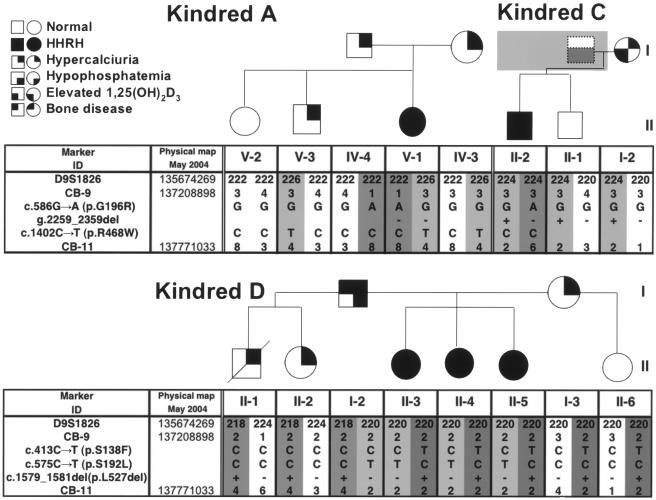
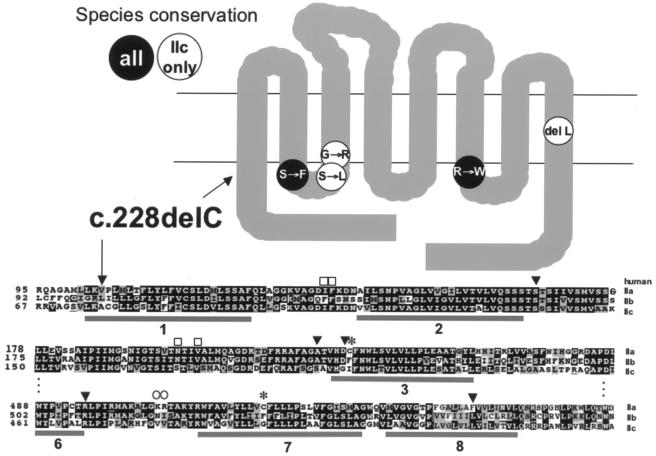
Hereditary hypophosphatemic rickets with hypercalciuria (HHRH) is a rare disorder of autosomal recessive inheritance that was first described in a large consanguineous Bedouin kindred. HHRH is characterized by the presence of hypophosphatemia secondary to renal phosphate wasting, radiographic and/or histological evidence of rickets, limb deformities, muscle weakness, and bone pain. HHRH is distinct from other forms of hypophosphatemic rickets in that affected individuals present with hypercalciuria due to increased serum 1,25-dihydroxyvitamin D levels and increased intestinal calcium absorption. We performed a genomewide linkage scan combined with homozygosity mapping, using genomic DNA from a large consanguineous Bedouin kindred that included 10 patients who received the diagnosis of HHRH. The disease mapped to a 1.6-Mbp region on chromosome 9q34, which contains SLC34A3, the gene encoding the renal sodium-phosphate cotransporter NaP(i)-IIc. Nucleotide sequence analysis revealed a homozygous single-nucleotide deletion (c.228delC) in this candidate gene in all individuals affected by HHRH. This mutation is predicted to truncate the NaP(i)-IIc protein in the first membrane-spanning domain and thus likely results in a complete loss of function of this protein in individuals homozygous for c.228delC. In addition, compound heterozygous missense and deletion mutations were found in three additional unrelated HHRH kindreds, which supports the conclusion that this disease is caused by SLC34A3 mutations affecting both alleles. Individuals of the investigated kindreds who were heterozygous for a SLC34A3 mutation frequently showed hypercalciuria, often in association with mild hypophosphatemia and/or elevations in 1,25-dihydroxyvitamin D levels. We conclude that NaP(i)-IIc has a key role in the regulation of phosphate homeostasis.
Figures
References
Web Resources
-
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/ (for SLC34A3 genomic contig [909158 in accession number NT_024000.16, or physical map nucleotide chr9:137401992 in human genome release March 2004], cDNA [187 in accession number NM_080877.1], and protein [accession number NP_543153.1]; SLC34A1 (NaPi-IIa) protein sequences for Homo sapiens [accession number NP_003043.2], Canis familiaris [accession number XP_536418.1], Pan troglodytes [accession number XP_518129.1], Rattus norvegicus [accession number NP_037162.1], Mus musculus [accession number NP_035522.1], and Gallus gallus [accession number XP_425204.1]; SLC34A2 (NaPi-IIb) protein sequences for H. sapiens [accession number NP_006415.1], C. familiaris [accession number XP_545968.1], P. troglodytes [accession number XP_526805.1], R. norvegicus [accession number XP_579555.1], M. musculus [accession number NP_035532.2], and G. gallus [accession number NP_989805.1]; and SLC34A3 (NaPi-IIc) protein sequences for C. familiaris [accession number XP_548353.1], R. norvegicus [accession number NP_647554.1], and M. musculus [accession number NP_543130.1])
-
- Marshfield Center for Medical Genetics, http://www2.marshfieldclinic.org/research/genetics/ (for Marshfield genetic maps)
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for HHRH, XLH, and ADHR)
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
