

SNARE-mediated lipid mixing depends on the physical state of the vesicles
- PMID: 16361343
- PMCID: PMC1386784
- DOI: 10.1529/biophysj.105.071415
SNARE-mediated lipid mixing depends on the physical state of the vesicles
Abstract
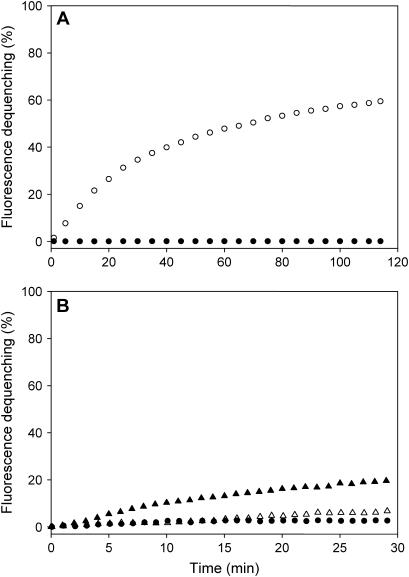
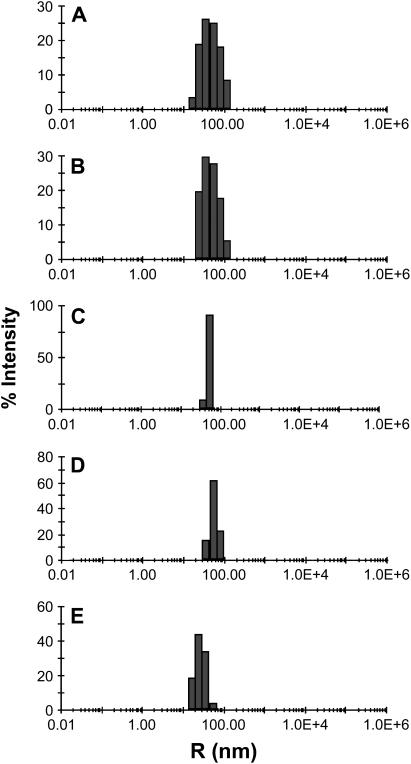
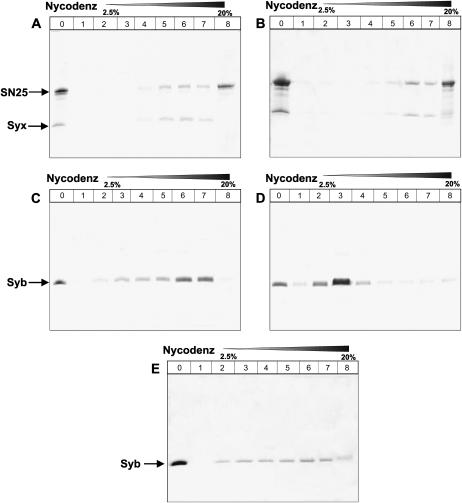
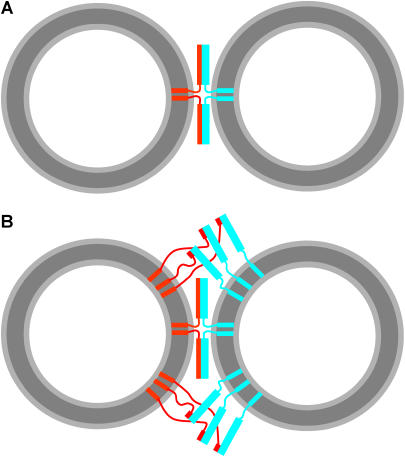
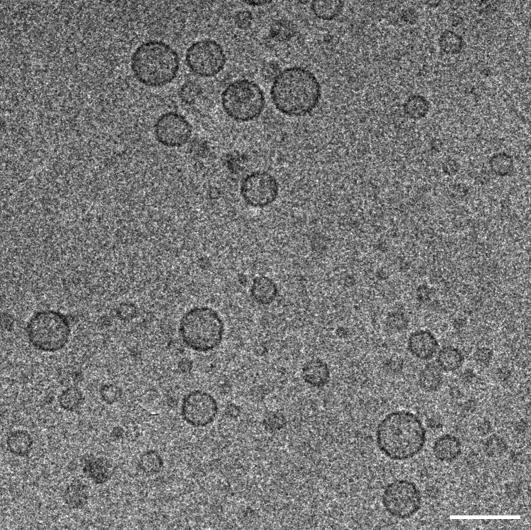
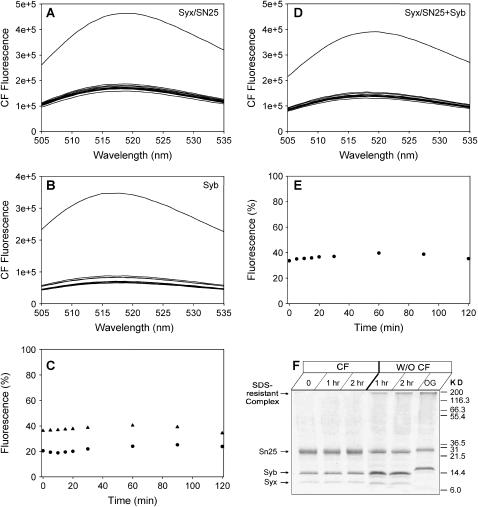
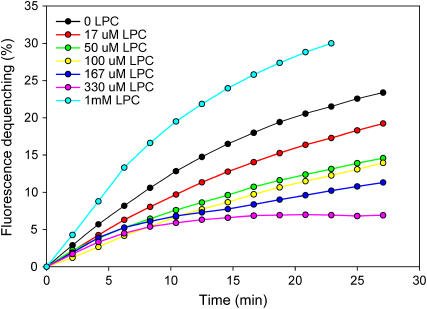
Reconstitution experiments have suggested that N-ethylmaleimide sensitive factor attachment protein receptor (SNARE) proteins constitute a minimal membrane fusion machinery but have yielded contradictory results, and it is unclear whether the mechanism of membrane merger is related to the stalk mechanism that underlies physiological membrane fusion. Here we show that reconstitution of solubilized neuronal SNAREs into preformed 100 nm liposomes (direct method) yields proteoliposomes with more homogeneous sizes and protein densities than the standard reconstitution method involving detergent cosolubilization of proteins and lipids. Standard reconstitutions yield slow but efficient lipid mixing at high protein densities and variable amounts of lipid mixing at moderate protein densities. However, the larger, more homogenous proteoliposomes prepared by the direct method yield almost no lipid mixing at moderate protein densities. These results suggest that the lipid mixing observed for standard reconstitutions is dominated by the physical state of the membrane, perhaps due to populations of small vesicles (or micelles) with high protein densities and curvature stress created upon reconstitution. Accordingly, changing membrane spontaneous curvature by adding lysophospholipids inhibits the lipid mixing observed for standard reconstitutions. Our data indicate that the lipid mixing caused by high SNARE densities and/or curvature stress occurs by a stalk mechanism resembling the mechanism of fusion between biological membranes, but the neuronal SNAREs are largely unable to induce lipid mixing at physiological protein densities and limited curvature stress.
Figures
References
-
- Pelham, H. R. 1999. SNAREs and the secretory pathway—lessons from yeast. Exp. Cell Res. 247:1–8. - PubMed
-
- Chen, Y. A., and R. H. Scheller. 2001. SNARE-mediated membrane fusion. Nat. Rev. Mol. Cell Biol. 2:98–106. - PubMed
-
- Rizo, J., and T. C. Sudhof. 2002. Snares and munc18 in synaptic vesicle fusion. Nat. Rev. Neurosci. 3:641–653. - PubMed
-
- Jahn, R., T. Lang, and T. C. Sudhof. 2003. Membrane fusion. Cell. 112:519–533. - PubMed
-
- Schiavo, G., M. Matteoli, and C. Montecucco. 2000. Neurotoxins affecting neuroexocytosis. Physiol. Rev. 80:717–766. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
