

Computer simulation of partitioning of ten pentapeptides Ace-WLXLL at the cyclohexane/water and phospholipid/water interfaces
- PMID: 16368010
- PMCID: PMC1351180
- DOI: 10.1186/1471-2091-6-30
Computer simulation of partitioning of ten pentapeptides Ace-WLXLL at the cyclohexane/water and phospholipid/water interfaces
Abstract
Background: Peptide-membrane interactions play a key role in the binding, partitioning and folding of membrane proteins, the activity of antimicrobial and fusion peptides, and a number of other processes. To gain a better understanding of the thermodynamics of such interactions, White and Wimley created an interfacial hydrophobicity scale based of the transfer free energy from water to octanol or lipid bilayers of a series of synthetic peptapeptides (Ace-WLXLL, with X being any of the twenty natural amino acids) (White and Wimley (1996) Nat. Struct. Biol. 3, 842-848). In this study, we performed molecular dynamics simulations of a representative set of ten of these peptides (X = D, K, R, N, A, T, S, I, F and W) in two membrane mimetic interfaces: water-cyclohexane (10 ns) and a fully solvated dioleoylphosphatidylcholine (DOPC) bilayer (50 ns) using both constant pressure and constant area ensembles. We focus on partitioning of the ten peptides at the cyclohexane/water and lipid/water interfaces.
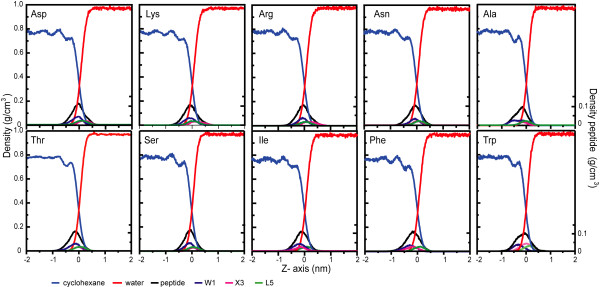
Results: The peptides rapidly equilibrate (< 2 ns) and partition at the cyclohexane/water interface. The X3 guest residue assumes average orientations that depend on the nature of the side chain. At the DOPC/water interface, dynamics is much slower and convergence is difficult to achieve on a 50 ns timescale. Nonetheless, all peptides partition to the lipid/water interface with distributions with widths of 1-2 nm. The peptides assume a broad range of side chain and backbone orientations and have only a small effect on the area of the unit cell. On average, hydrophobic guest residues partition deeper into the hydrophobic core than hydrophilic residues. In some cases the peptides penetrate sufficiently deep to somewhat affect the distribution of the C=C double bond in DOPC. The relative distribution of the X3 guest residue compared to W1 and L5 is similar in the water/cyclohexane and water/lipid simulations. Snapshots show mostly extended backbone conformations in both environments. There is little difference between simulations at a constant area of 0.66 nm2 and simulations at constant pressure that approximately yield the same average area of 0.66 nm2.
Conclusion: These peptides were designed to assume extended conformations, which is confirmed by the simulations. The distribution of the X3 side chain depends on its nature, and can be determined from molecular dynamics simulations. The time scale of peptide motion at a phospholipids-water interface is too long to directly calculate the experimentally measured hydrophobicity scale to test and improve the simulation parameters. This should be possible at the water/cyclohexane interface and likely will become feasible in the future for the phospholipids/water case.
Figures










References
-
- Kessel A, Ben-Tal N. Free energy determinants of peptide association with lipids bilayers. Current Topics in Membranes. 2002;52:205–253.
-
- Epand RM. Fusion peptides and the mechanism of viral fusion. Biochim Biophys Acta. 2003;1614:116–121. - PubMed
-
- Epand RM, Vogel HJ. Diversity of antimicrobial peptides and their mechanisms of action. Biochim Biophys Acta. 1999;1462:11–28. - PubMed
-
- Murray D, Arbuzova A, Honig B, MacLaughlin S. The role of electrostatic and nonpolar interactions in the association of peripheral protiens with membranes. Peptide-lipid interactions. 2002;52:277–307.
-
- Wimley WC, White SH. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat Struct Biol. 1996;3:842–848. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
