

Histone deacetylase 2-mediated deacetylation of the glucocorticoid receptor enables NF-kappaB suppression
- PMID: 16380507
- PMCID: PMC2118081
- DOI: 10.1084/jem.20050466
Histone deacetylase 2-mediated deacetylation of the glucocorticoid receptor enables NF-kappaB suppression
Abstract
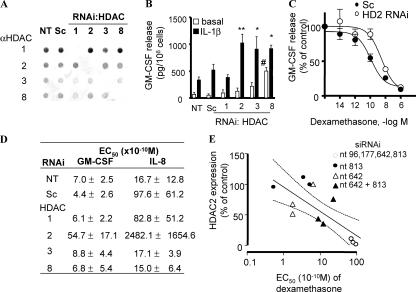
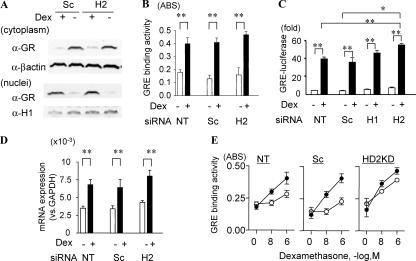
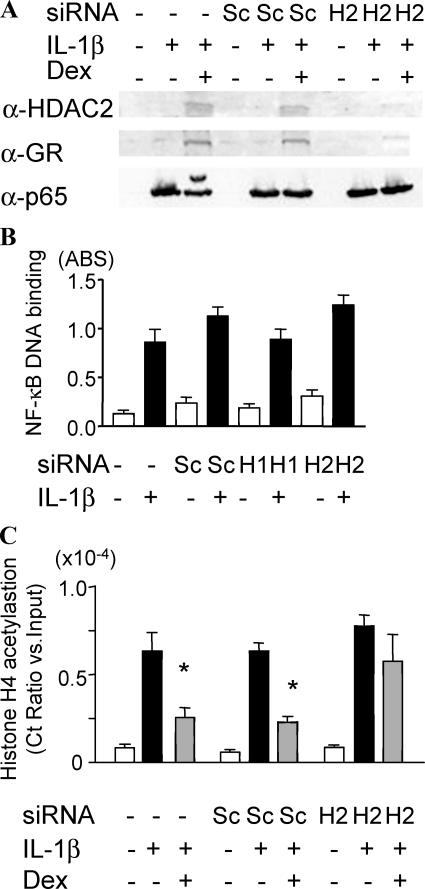
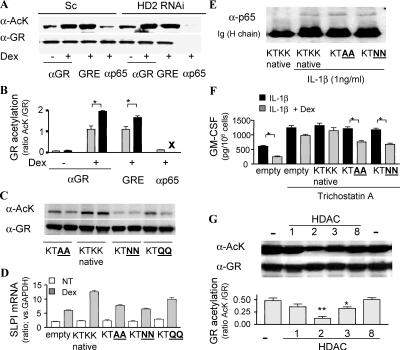
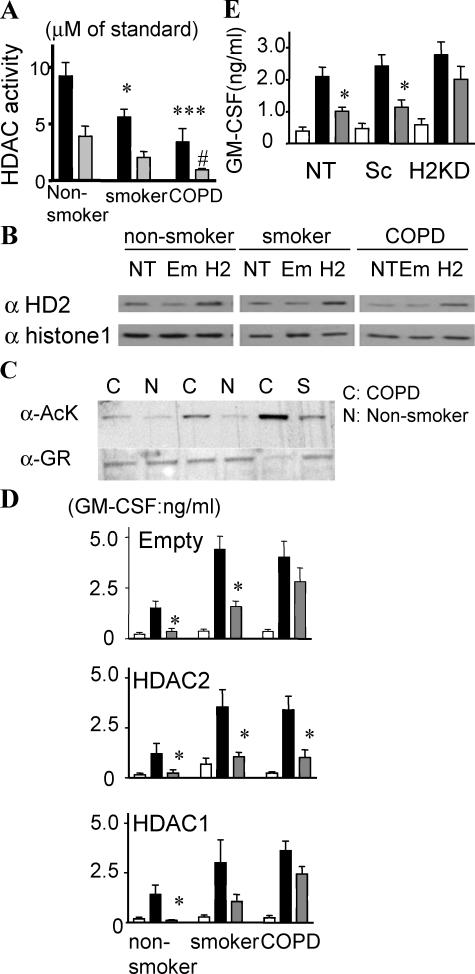
Glucocorticoids are the most effective antiinflammatory agents for the treatment of chronic inflammatory diseases even though some diseases, such as chronic obstructive pulmonary disease (COPD), are relatively glucocorticoid insensitive. However, the molecular mechanism of this glucocorticoid insensitivity remains uncertain. We show that a defect of glucocorticoid receptor (GR) deacetylation caused by impaired histone deacetylase (HDAC) 2 induces glucocorticoid insensitivity toward nuclear factor (NF)-kappaB-mediated gene expression. Specific knockdown of HDAC2 by RNA interference resulted in reduced sensitivity to dexamethasone suppression of interleukin 1beta-induced granulocyte/macrophage colony-stimulating factor production. Loss of HDAC2 did not reduce GR nuclear translocation, GR binding to glucocorticoid response element (GRE) on DNA, or GR-induced DNA or gene induction but inhibited the association between GR and NF-kappaB. GR becomes acetylated after ligand binding, and HDAC2-mediated GR deacetylation enables GR binding to the NF-kappaB complex. Site-directed mutagenesis of K494 and K495 reduced GR acetylation, and the ability to repress NF-kappaB-dependent gene expression becomes insensitive to histone deacetylase inhibition. In conclusion, we show that overexpression of HDAC2 in glucocorticoid-insensitive alveolar macrophages from patients with COPD is able to restore glucocorticoid sensitivity. Thus, reduction of HDAC2 plays a critical role in glucocorticoid insensitivity in repressing NF-kappaB-mediated, but not GRE-mediated, gene expression.
Figures
References
-
- Beato, M. 1996. Chromatin structure and the regulation of gene expression: remodeling at the MMTV promoter. J. Mol. Med. 74:711–724. - PubMed
-
- Wolffe, A.P. 1997. Transcriptional control. Sinful repression. Nature. 387:16–17. - PubMed
-
- Thiagalingam, S., K.H. Cheng, H.J. Lee, N. Mineva, A. Thiagalingam, and J.F. Ponte. 2003. Histone deacetylases: unique players in shaping the epigenetic histone code. Ann. NY Acad. Sci. 983:84–100. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
