

A homozygous missense mutation in TGM5 abolishes epidermal transglutaminase 5 activity and causes acral peeling skin syndrome
- PMID: 16380904
- PMCID: PMC1285176
- DOI: 10.1086/497707
A homozygous missense mutation in TGM5 abolishes epidermal transglutaminase 5 activity and causes acral peeling skin syndrome
Abstract
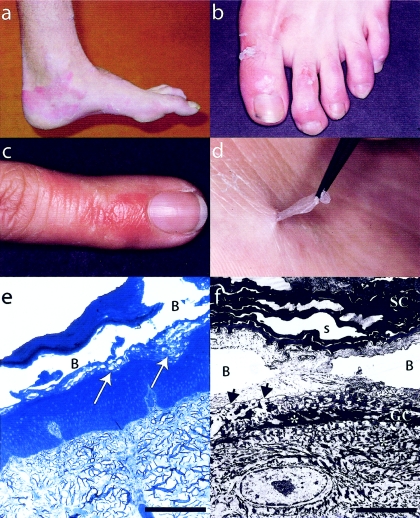
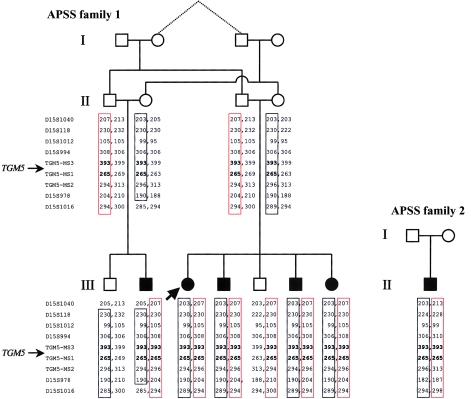
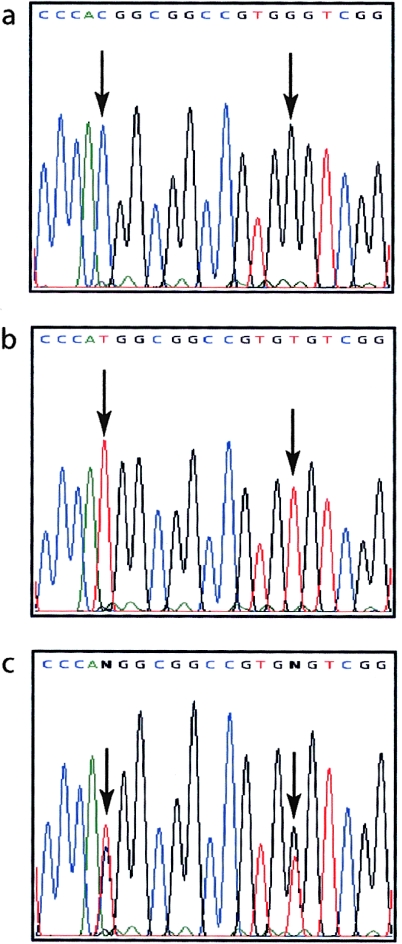
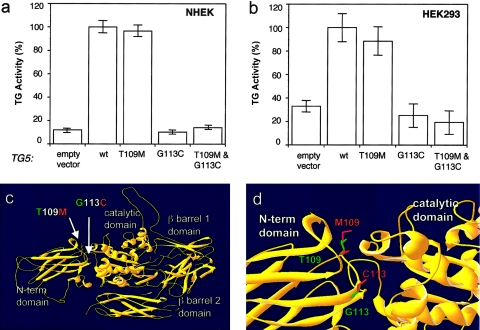
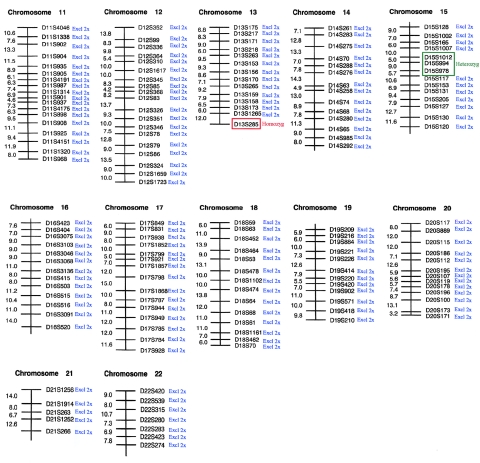
Peeling skin syndrome is an autosomal recessive genodermatosis characterized by the shedding of the outer epidermis. In the acral form, the dorsa of the hands and feet are predominantly affected. Ultrastructural analysis has revealed tissue separation at the junction between the granular cells and the stratum corneum in the outer epidermis. Genomewide linkage analysis in a consanguineous Dutch kindred mapped the gene to 15q15.2 in the interval between markers D15S1040 and D15S1016. Two homozygous missense mutations, T109M and G113C, were found in TGM5, which encodes transglutaminase 5 (TG5), in all affected persons in two unrelated families. The mutation was present on the same haplotype in both kindreds, indicating a probable ancestral mutation. TG5 is strongly expressed in the epidermal granular cells, where it cross-links a variety of structural proteins in the terminal differentiation of the epidermis to form the cornified cell envelope. An established, in vitro, biochemical cross-linking assay revealed that, although T109M is not pathogenic, G113C completely abolishes TG5 activity. Three-dimensional modeling of TG5 showed that G113C lies close to the catalytic domain, and, furthermore, that this glycine residue is conserved in all known transglutaminases, which is consistent with pathogenicity. Other families with more-widespread peeling skin phenotypes lacked TGM5 mutations. This study identifies the first causative gene in this heterogeneous group of skin disorders and demonstrates that the protein cross-linking function performed by TG5 is vital for maintaining cell-cell adhesion between the outermost layers of the epidermis.
Figures

References
Web Resources
-
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/ (for TGM5 isoform 1 [accession numbers NM_201631.1 and AF035961] and isoform 2 [accession numbers NM_004245.1 and AF035960])
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for PSS and spherocytic elliptocytosis)
-
- Protein Data Bank, http://www.rscb.org/pdb/ (for crystal structure of human TG2 [identification number 1KV3])
-
- UCSC Genome Browser, http://www.genome.ucsc.edu/ (for TGM5, TGM7, and EPB42)
References
-
- Abdel-Hafez K, Safer AM, Selim MM, Rehak A (1983) Familial continual skin peeling. Dermatologica 166:23–31 - PubMed
-
- Bouhassira EE, Schwartz RS, Yawata Y, Ata K, Kanzaki A, Qiu JJ, Nagel RL, Rybicki AC (1992) An alanine-to-threonine substitution in protein 4.2 cDNA is associated with a Japanese form of hereditary hemolytic anemia (protein 4.2NIPPON). Blood 79:1846–1854 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
