

The Database of Macromolecular Motions: new features added at the decade mark
- PMID: 16381870
- PMCID: PMC1347409
- DOI: 10.1093/nar/gkj046
The Database of Macromolecular Motions: new features added at the decade mark
Abstract
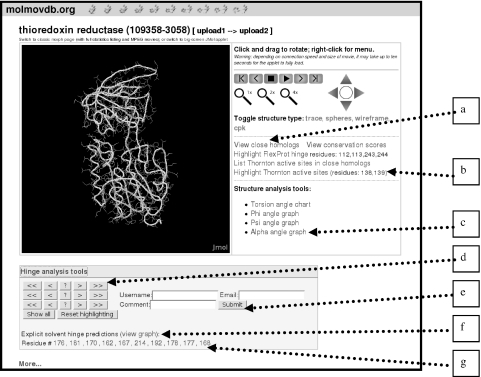
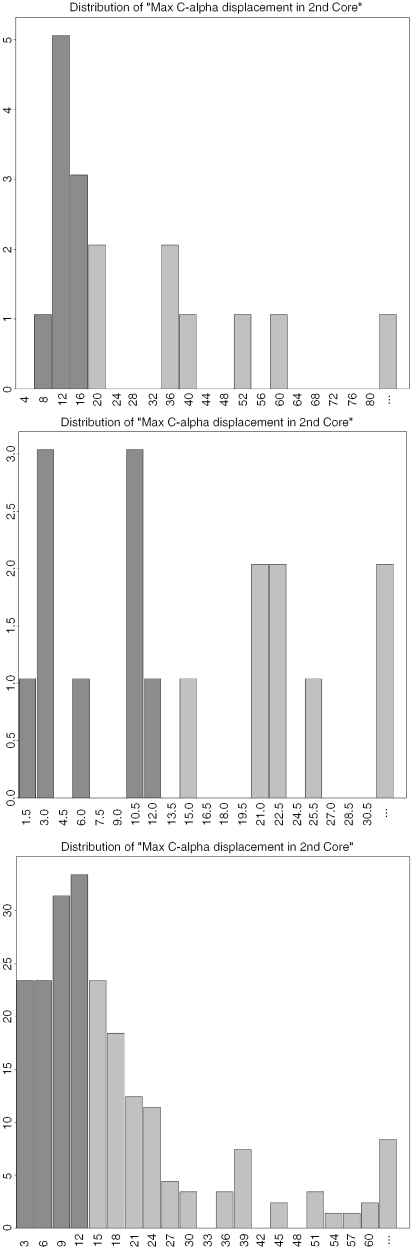
The database of molecular motions, MolMovDB (http://molmovdb.org), has been in existence for the past decade. It classifies macromolecular motions and provides tools to interpolate between two conformations (the Morph Server) and predict possible motions in a single structure. In 2005, we expanded the services offered on MolMovDB. In particular, we further developed the Morph Server to produce improved interpolations between two submitted structures. We added support for multiple chains to the original adiabatic mapping interpolation, allowing the analysis of subunit motions. We also added the option of using FRODA interpolation, which allows for more complex pathways, potentially overcoming steric barriers. We added an interface to a hinge prediction service, which acts on single structures and predicts likely residue points for flexibility. We developed tools to relate such points of flexibility in a structure to particular key residue positions, i.e. active sites or highly conserved positions. Lastly, we began relating our motion classification scheme to function using descriptions from the Gene Ontology Consortium.
Figures
Similar articles
-
Normal mode analysis of macromolecular motions in a database framework: developing mode concentration as a useful classifying statistic.Proteins. 2002 Sep 1;48(4):682-95. doi: 10.1002/prot.10168. Proteins. 2002. PMID: 12211036
-
RigidFinder: a fast and sensitive method to detect rigid blocks in large macromolecular complexes.Proteins. 2010 Feb 1;78(2):309-24. doi: 10.1002/prot.22544. Proteins. 2010. PMID: 19705487
-
ProSAT2--Protein Structure Annotation Server.Nucleic Acids Res. 2006 Jul 1;34(Web Server issue):W79-83. doi: 10.1093/nar/gkl216. Nucleic Acids Res. 2006. PMID: 16845114 Free PMC article.
-
High-throughput modeling and analysis of protein structural dynamics.Brief Bioinform. 2007 Nov;8(6):432-45. doi: 10.1093/bib/bbm014. Epub 2007 May 7. Brief Bioinform. 2007. PMID: 17485424 Review.
-
Macromolecular modeling and design in Rosetta: recent methods and frameworks.Nat Methods. 2020 Jul;17(7):665-680. doi: 10.1038/s41592-020-0848-2. Epub 2020 Jun 1. Nat Methods. 2020. PMID: 32483333 Free PMC article. Review.
Cited by
-
Normal Mode Analysis as a Routine Part of a Structural Investigation.Molecules. 2019 Sep 10;24(18):3293. doi: 10.3390/molecules24183293. Molecules. 2019. PMID: 31510014 Free PMC article. Review.
-
How well can we understand large-scale protein motions using normal modes of elastic network models?Biophys J. 2007 Aug 1;93(3):920-9. doi: 10.1529/biophysj.106.095927. Epub 2007 May 4. Biophys J. 2007. PMID: 17483178 Free PMC article.
-
Identifying the minimal sets of distance restraints for FRET-assisted protein structural modeling.ArXiv [Preprint]. 2024 Aug 19:arXiv:2405.07983v2. ArXiv. 2024. Update in: Protein Sci. 2024 Dec;33(12):e5219. doi: 10.1002/pro.5219. PMID: 38800659 Free PMC article. Updated. Preprint.
-
StoneHinge: hinge prediction by network analysis of individual protein structures.Protein Sci. 2009 Feb;18(2):359-71. doi: 10.1002/pro.38. Protein Sci. 2009. PMID: 19180449 Free PMC article.
-
Principles and Overview of Sampling Methods for Modeling Macromolecular Structure and Dynamics.PLoS Comput Biol. 2016 Apr 28;12(4):e1004619. doi: 10.1371/journal.pcbi.1004619. eCollection 2016 Apr. PLoS Comput Biol. 2016. PMID: 27124275 Free PMC article. Review.
References
-
- Krebs W.G., Alexandrov V., Wilson C.A., Echols N., Yu H., Gerstein M. Normal mode analysis of macromolecular motions in a database framework: developing mode concentration as a useful classifying statistic. Proteins. 2002;48:682–695. - PubMed
-
- Krebs W.G., Tsai J., Alexandrov V., Junker J., Jansen R., Gerstein M. Tools and databases to analyze protein flexibility; approaches to mapping implied features onto sequences. Methods Enzymol. 2003;374:544–584. - PubMed
-
- Qian J., Stenger B., Wilson C.A., Lin J., Jansen R., Teichmann S.A., Park J., Krebs W.G., Yu H., Alexandrov V., et al. PartsList: a web-based system for dynamically ranking protein folds based on disparate attributes, including whole-genome expression and interaction information. Nucleic Acids Res. 2001;29:1750–1764. - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
