

Cystic fibrosis: terminology and diagnostic algorithms
- PMID: 16384879
- PMCID: PMC2104676
- DOI: 10.1136/thx.2005.043539
Cystic fibrosis: terminology and diagnostic algorithms
Abstract
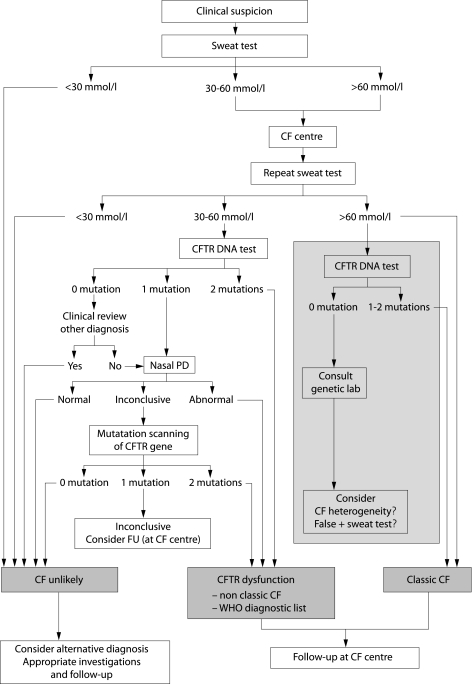
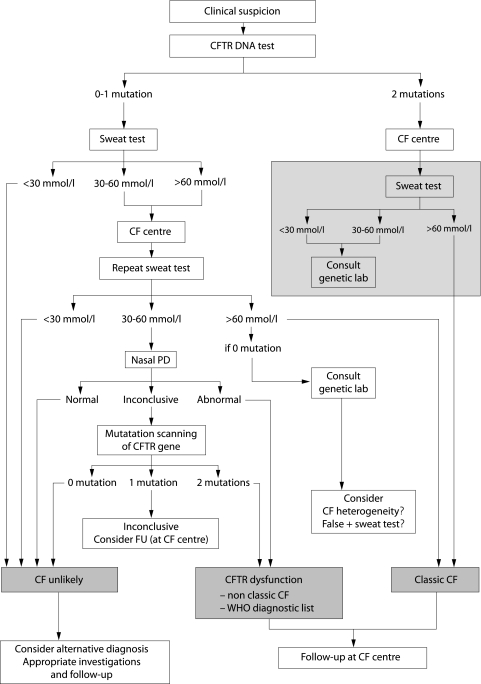
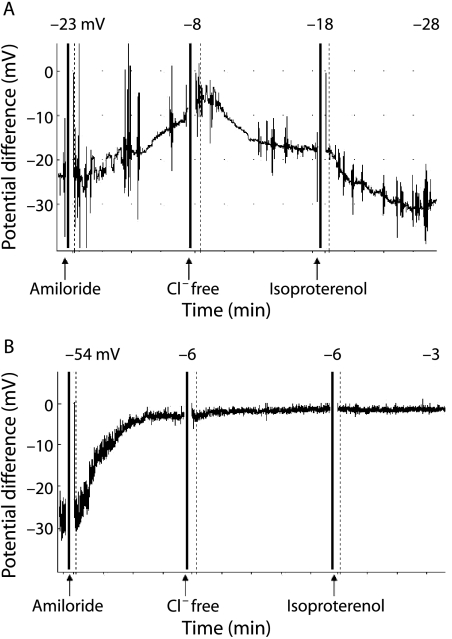
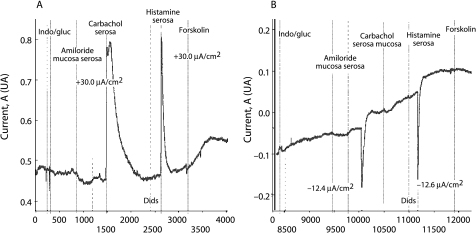
There is great heterogeneity in the clinical manifestations of cystic fibrosis (CF). Some patients may have all the classical manifestations of CF from infancy and have a relatively poor prognosis, while others have much milder or even atypical disease manifestations and still carry mutations on each of the CFTR genes. It is important to distinguish between these categories of patients. The European Diagnostic Working Group proposes the following terminology. Patients are diagnosed with classic or typical CF if they have one or more phenotypic characteristics and a sweat chloride concentration of >60 mmol/l. The vast majority of CF patients fall into this category. Usually one established mutation causing CF can be identified on each CFTR gene. Patients with classic CF can have exocrine pancreatic insufficiency or pancreatic sufficiency. The disease can have a severe course with rapid progression of symptoms or a milder course with very little deterioration over time. Patients with non-classic or atypical CF have a CF phenotype in at least one organ system and a normal (<30 mmol/l) or borderline (30-60 mmol/l) sweat chloride level. In these patients confirmation of the diagnosis of CF requires detection of one disease causing mutation on each CFTR gene or direct quantification of CFTR dysfunction by nasal potential difference measurement. Non-classic CF includes patients with multiorgan or single organ involvement. Most of these patients have exocrine pancreatic sufficiency and milder lung disease. Algorithms for a structured diagnostic process are proposed.
Conflict of interest statement
Competing interests: none declared.
Comment in
-
Diagnosing CF: sweat, blood and years.Thorax. 2006 Jul;61(7):556-7. doi: 10.1136/thx.2005.056309. Thorax. 2006. PMID: 16807389 Free PMC article.
-
Sweat testing in CF.Thorax. 2007 May;62(5):462; author reply 463. doi: 10.1136/thx.2006.070946. Thorax. 2007. PMID: 17468461 Free PMC article. No abstract available.
References
-
- Orenstein B M, Winnie G B, Altman H. Cystic fibrosis: a 2002 update. J Pediatr 2002140156–164. - PubMed
-
- World Health Organization Classification of cystic fibrosis and related disorders. Report of a Joint Working Group of WHO/ICF(M)A/ECFS/ECFTN, 2001 (reprinted in J Cyst Fibros 200215–8.
-
- Goss C H, Rosenfeld M. Update on cystic fibrosis epidemiology. Curr Opin Pulm Med 200410510–514. - PubMed
-
- Boyle M P. Nonclassic cystic fibrosis and CFTR‐related diseases. Curr Opin Pulm Med 20039498–503. - PubMed
-
- Rosenstein B J, Cutting G R, for the Cystic Fibrosis Foundation Consensus Panel The diagnosis of cystic fibrosis: a consensus statement. J Pediatr 1998132589–595. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical