

Dissection of the bacteriophage Mu strong gyrase site (SGS): significance of the SGS right arm in Mu biology and DNA gyrase mechanism
- PMID: 16385052
- PMCID: PMC1347280
- DOI: 10.1128/JB.188.2.619-632.2006
Dissection of the bacteriophage Mu strong gyrase site (SGS): significance of the SGS right arm in Mu biology and DNA gyrase mechanism
Abstract
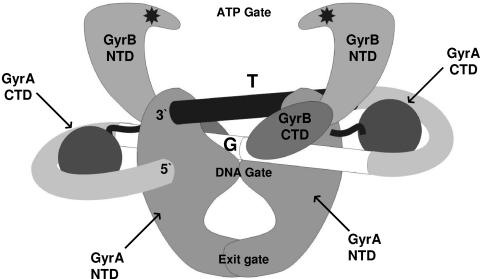
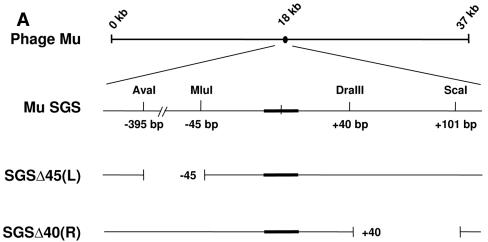
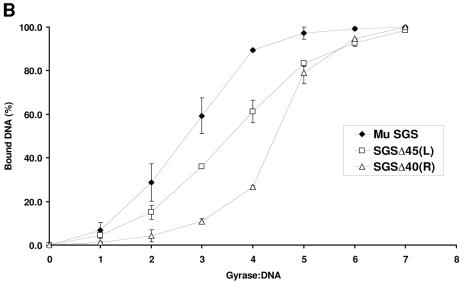
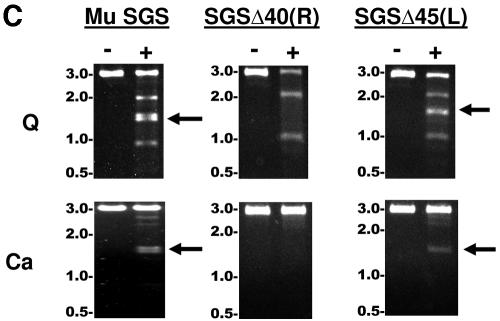
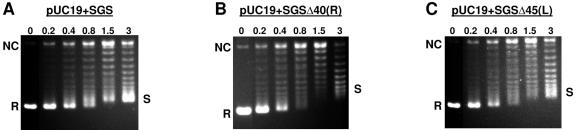
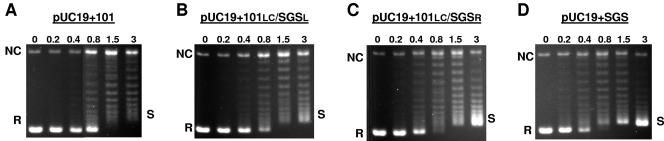
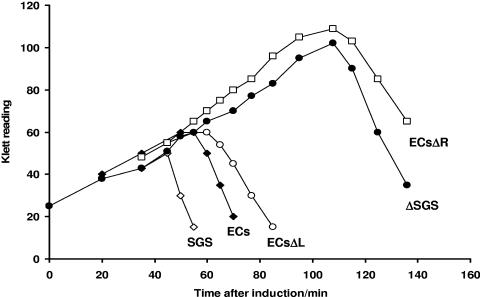
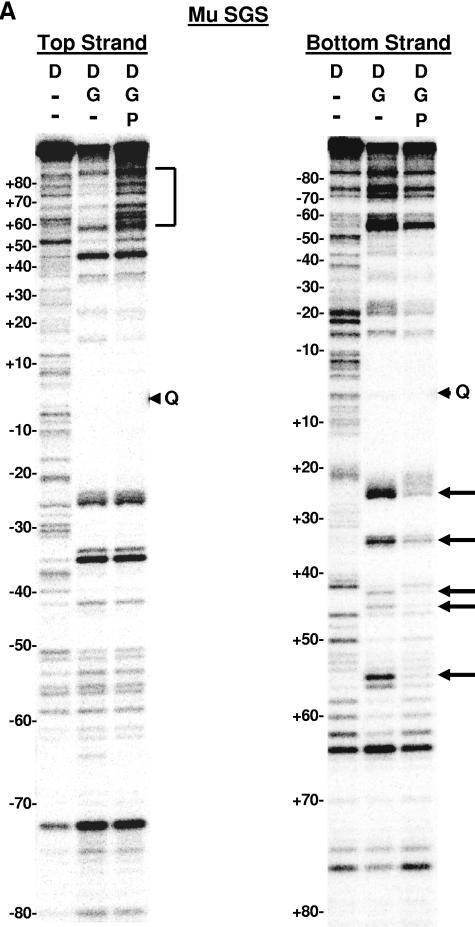
The bacteriophage Mu strong gyrase site (SGS), required for efficient phage DNA replication, differs from other gyrase sites in the efficiency of gyrase binding coupled with a highly processive supercoiling activity. Genetic studies have implicated the right arm of the SGS as a key structural feature for promoting rapid Mu replication. Here, we show that deletion of the distal portion of the right arm abolishes efficient binding, cleavage, and supercoiling by DNA gyrase in vitro. DNase I footprinting analysis of the intact SGS revealed an adenylyl imidodiphosphate-dependent change in protection in the right arm, indicating that this arm likely forms the T segment that is passed through the cleaved G segment during the supercoiling reaction. Furthermore, in an SGS derivative with an altered right-arm sequence, the left arm showed these changes, suggesting that the selection of a T segment by gyrase is determined primarily by the sequences of the arms. Analysis of the sequences of the SGS and other gyrase sites suggests that the choice of T segment correlates with which arm possesses the more extensive set of phased anisotropic bending signals, with the Mu right arm possessing an unusually extended set of such signals. The implications of these observations for the structure of the gyrase-DNA complex and for the biological function of the Mu SGS are discussed.
Figures
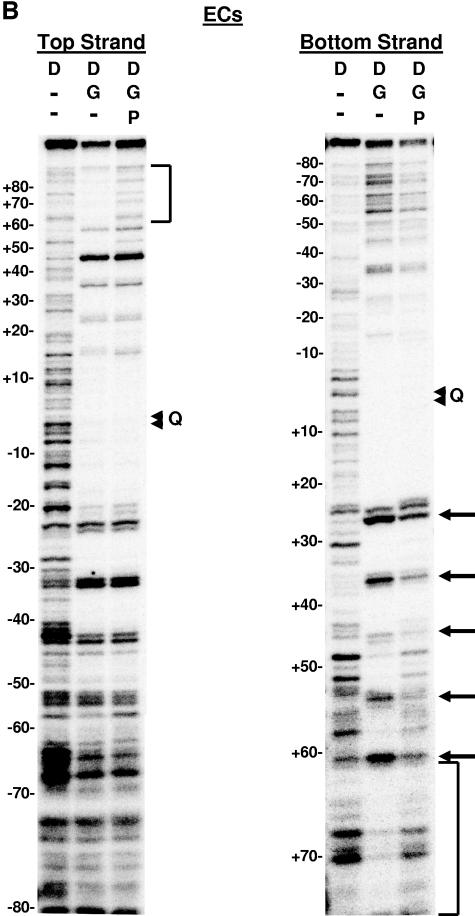
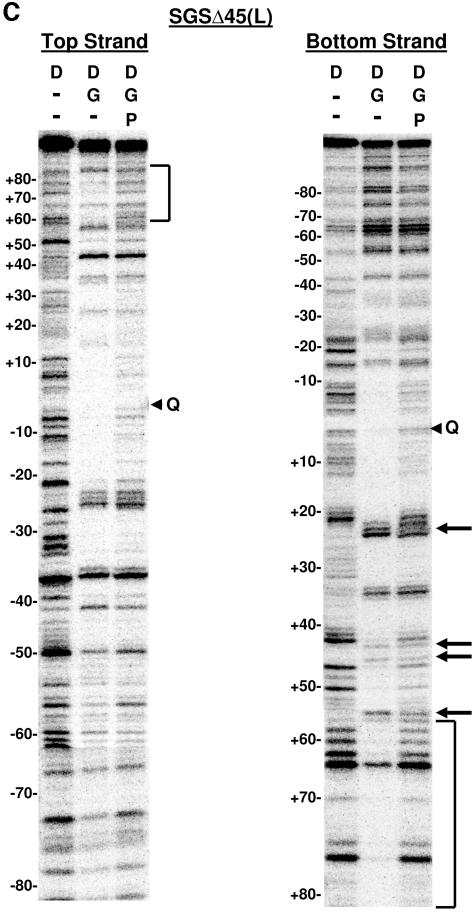
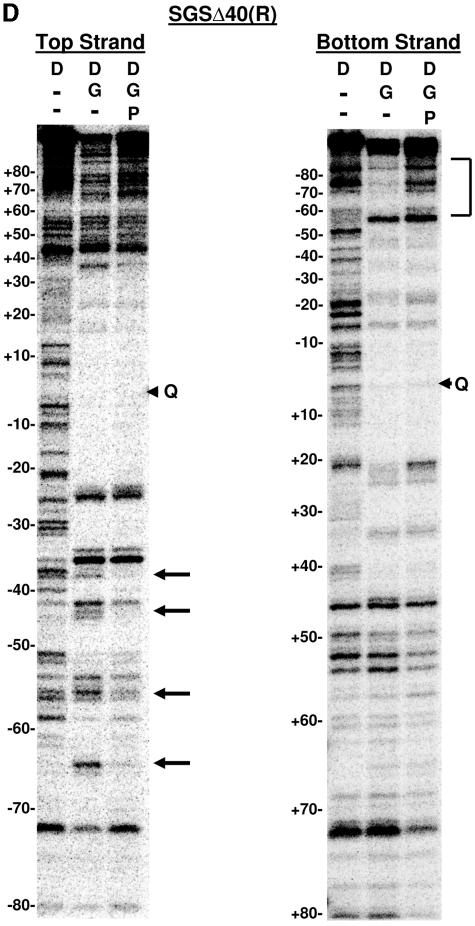
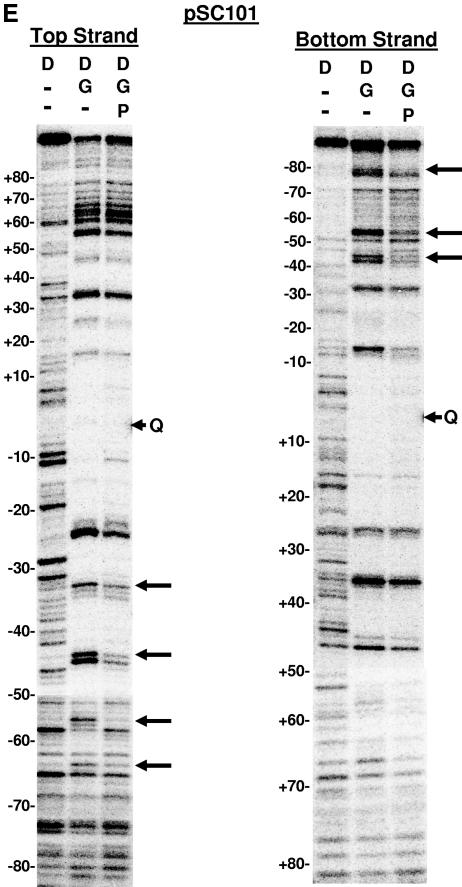
References
-
- Berger, J. M., S. J. Gamblin, S. C. Harrison, and J. C. Wang. 1996. Structure and mechanism of DNA topoisomerase II. Nature 379:225-232. - PubMed
-
- Champoux, J. J. 2001. DNA topoisomerases: structure, function, and mechanism. Annu. Rev. Biochem. 70:369-413. - PubMed
-
- Costenaro, L., J. G. Grossmann, C. Ebel, and A. Maxwell. 2005. Small-angle X-ray scattering reveals the solution structure of the full-length DNA gyrase a subunit. Structure (Cambridge) 13:287-296. - PubMed
-
- Craigie, R., and K. Mizuuchi. 1986. Role of DNA topology in Mu transposition: mechanism of sensing the relative orientation of two DNA segments. Cell 45:793-800. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
