

Contribution of a common single-nucleotide polymorphism to the genetic predisposition for erythropoietic protoporphyria
- PMID: 16385445
- PMCID: PMC1380220
- DOI: 10.1086/498620
Contribution of a common single-nucleotide polymorphism to the genetic predisposition for erythropoietic protoporphyria
Abstract
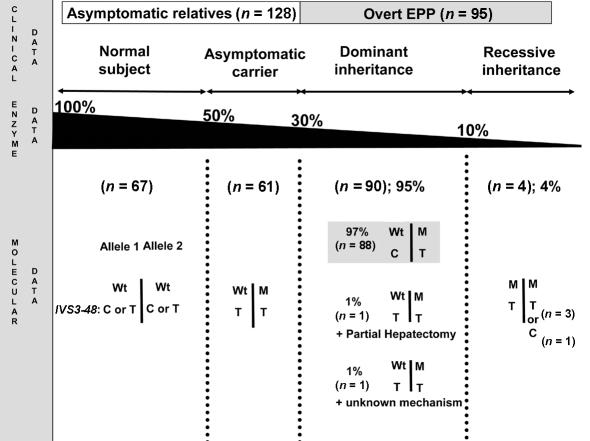
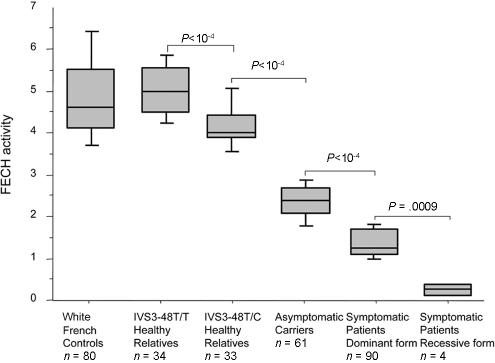
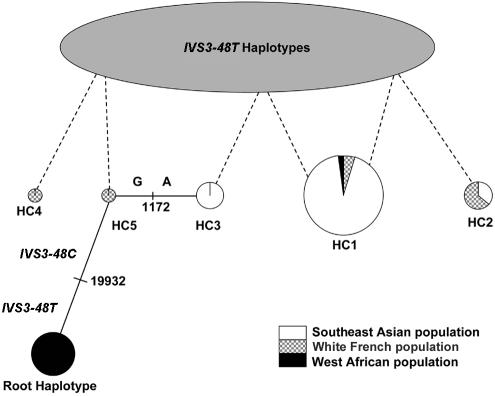
Erythropoietic protoporphyria (EPP) is an inherited disorder of heme biosynthesis that results from a partial deficiency of ferrochelatase (FECH). Recently, we have shown that the inheritance of the common hypomorphic IVS3-48C allele trans to a deleterious mutation reduces FECH activity to below a critical threshold and accounts for the photosensitivity seen in patients. Rare cases of autosomal recessive inheritance have been reported. We studied a cohort of 173 white French EPP families and a group of 360 unrelated healthy subjects from four ethnic groups. The prevalences of the recessive and dominant autosomal forms of EPP are 4% (95% confidence interval 1-8) and 95% (95% confidence interval 91-99), respectively. In 97.9% of dominant cases, an IVS3-48C allele is co-inherited with the deleterious mutation. The frequency of the IVS3-48C allele differs widely in the Japanese (43%), southeast Asian (31%), white French (11%), North African (2.7%), and black West African (<1%) populations. These differences can be related to the prevalence of EPP in these populations and could account for the absence of EPP in black subjects. The phylogenic origin of the IVS3-48C haplotypes strongly suggests that the IVS3-48C allele arose from a single recent mutational event. Estimation of the age of the IVS3-48C allele from haplotype data in white and Asian populations yields an estimated age three to four times younger in the Japanese than in the white population, and this difference may be attributable either to differing demographic histories or to positive selection for the IVS3-48C allele in the Asian population. Finally, by calculating the KA/KS ratio in humans and chimpanzees, we show that the FECH protein sequence is subject to strong negative pressure. Overall, EPP looks like a Mendelian disorder, in which the prevalence of overt disease depends mainly on the frequency of a single common single-nucleotide polymorphism resulting from a unique mutational event that occurred 60,000 years ago.
Figures

References
Web Resources
-
- ARLEQUIN, http://anthropologie.unige.ch/arlequin/
-
- GenBank, http://www.ncbi.nlm.nih.gov/GenBank/ (for chimpanzee FECH sequence [DQ149645], human FECH cDNA [NM000140], human FECH gene [AJ250235], and human FECH protein [NP000131]
-
- Human Gene Mutation Database (HGMD), http://archive.uwcm.ac.uk/uwcm/mg/hgmd0.html/
-
- Human Genome Center, Institute of Medical Science, University of Tokyo, http://www.hgc.ims.u-tokyo.ac.jp/
References
-
- Anderson KE, Sassa S, Bishop DF, Desnick RJ (2001) Disorders of heme biosynthesis: X-linked sideroblastic anemia and the porphyrias. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The metabolic and molecular basis of inherited disease. 8th ed. McGraw-Hill, New York, pp 2961–3062
-
- Bloomer JR (1988) The liver in protoporphyria. Hepatology 8:402–407 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
