

Comparative Study
doi: 10.1086/499164.
Epub 2005 Nov 21.
Mutations in TRIOBP, which encodes a putative cytoskeletal-organizing protein, are associated with nonsyndromic recessive deafness
Affiliations
- PMID: 16385457
- PMCID: PMC1380211
- DOI: 10.1086/499164
Item in Clipboard
Comparative Study
Mutations in TRIOBP, which encodes a putative cytoskeletal-organizing protein, are associated with nonsyndromic recessive deafness
Am J Hum Genet.
2006 Jan.
Abstract
In seven families, six different mutant alleles of TRIOBP on chromosome 22q13 cosegregate with autosomal recessive nonsyndromic deafness. These alleles include four nonsense (Q297X, R788X, R1068X, and R1117X) and two frameshift (D1069fsX1082 and R1078fsX1083) mutations, all located in exon 6 of TRIOBP. There are several alternative splice isoforms of this gene, the longest of which, TRIOBP-6, comprises 23 exons. The linkage interval for the deafness segregating in these families includes DFNB28. Genetic heterogeneity at this locus is suggested by three additional families that show significant evidence of linkage of deafness to markers on chromosome 22q13 but that apparently have no mutations in the TRIOBP gene.
Figures
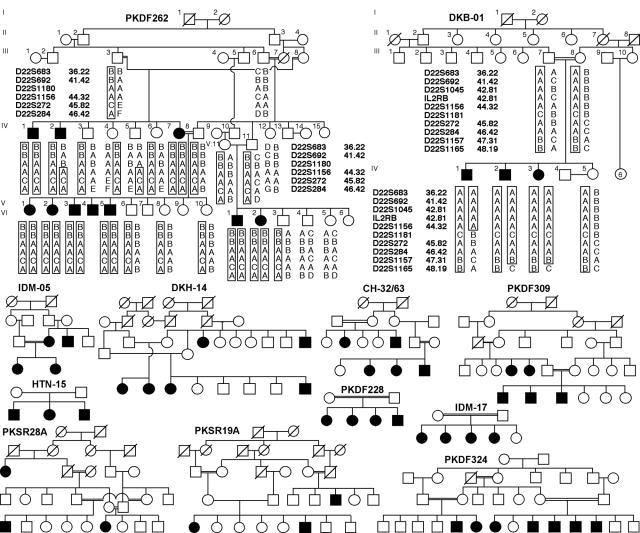
Pedigrees of 12 consanguineous families from Pakistan and India. Blackened symbols represent individuals with clinically documented congenital deafness. Genotype and haplotype data for families PKDF262 and DKB-01 are shown. For the chromosome 22q13 locus, the haplotype of affected individual IV:2 of family PKDF262 has a proximal breakpoint at D22S1180. Affected individual IV:1 of family DKB-01 has a distal breakpoint at D22S1181, which refines the smallest linkage interval to 702 kb. However, when TRIOBP mutation–containing families are removed, the minimal deafness-linkage interval is ∼10 cM, defined by two of the remaining families, PKSR19A and PKSR28A, with simulated LOD scores of 3.4 and 3.1, respectively (table 1).
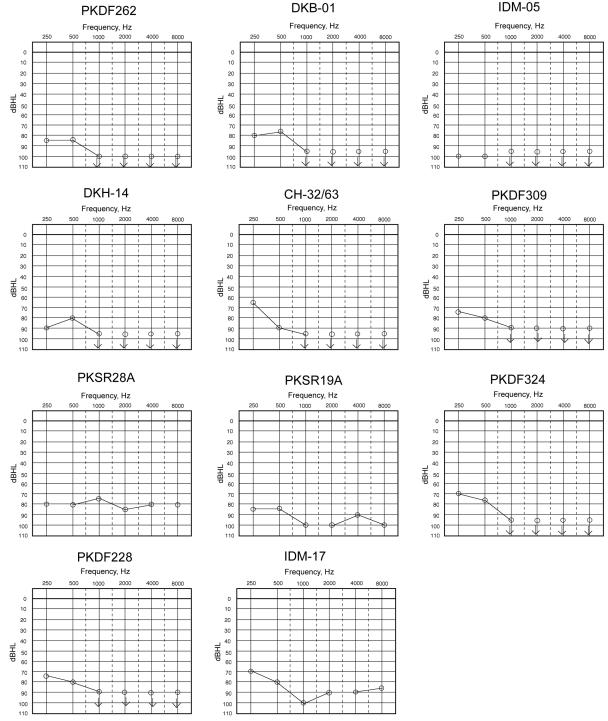
Audiograms of one congenitally deaf individual from each family. The carriers of a mutant allele have normal hearing.
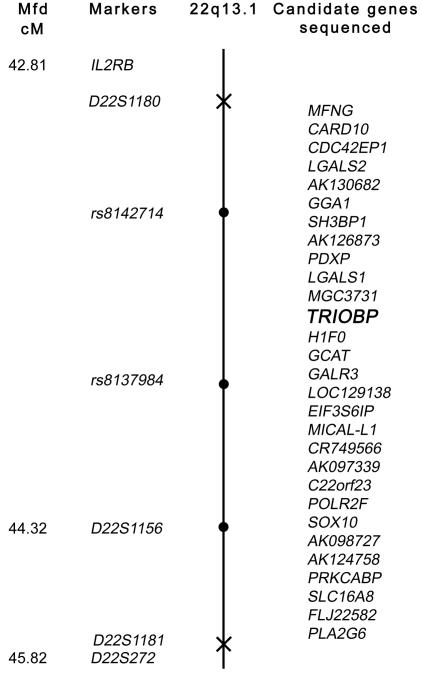
Genetic map of the 22q13 locus defined by proximal and distal meiotic breakpoints in families PKDF262 and DKB-01, respectively. The Marshfield map distance (Mfd) of polymorphic markers is shown (left) (see Center for Medical Genetics, Marshfield Medical Research Foundation Web site). The proximal breakpoint, D22S1180, and the distal breakpoint, D22S1181, are marked with an X on the chromosome. Linked markers are shown as blackened circles on the chromosome. In two affected individuals from at least six of the families, we evaluated the other 28 annotated genes in this interval for mutations but found none. TRIOBP is shown in bold italics.
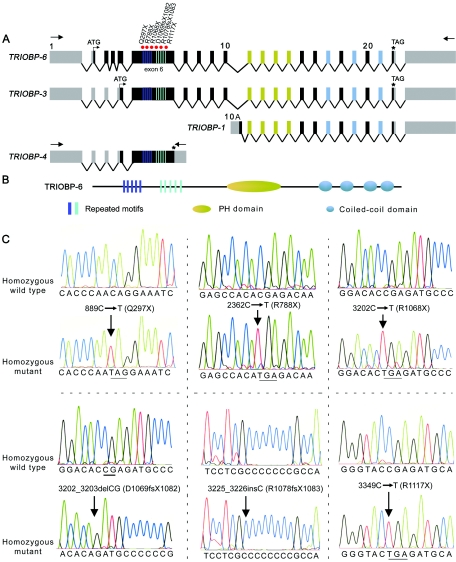
Structure and mutations of TRIOBP associated with deafness. A, Schematic representation of the genomic structure of isoforms of human TRIOBP and the locations of the mutations in exon 6 associated with deafness. Arrows represent the location of the primers used for RT-PCR. Red dots indicate the site of the mutations in exon 6 of TRIOBP. B, Protein structure of TRIOBP-6. There are five copies each of two different repeated motifs (dark blue and light blue rectangles) that have the amino acid sequences TPCA/I/TQR/WDNPRASSPNRT/ST/AQRDN/SPR and VCIGHRDAPRAS/TS/FPP. C, Mutations of TRIOBP segregating in six families. Electropherograms of amplimers from genomic DNA templates illustrate homozygosity for mutations found in all affected family members and homozygosity for the wild-type allele in an unaffected individual. All obligate carriers are heterozygous (not shown). All of the mutations described here are numbered beginning with +1 at the A base of the translation start codon (ATG) in exon 2 of TRIOBP-6. The stop codons due to nonsense mutations are underlined.
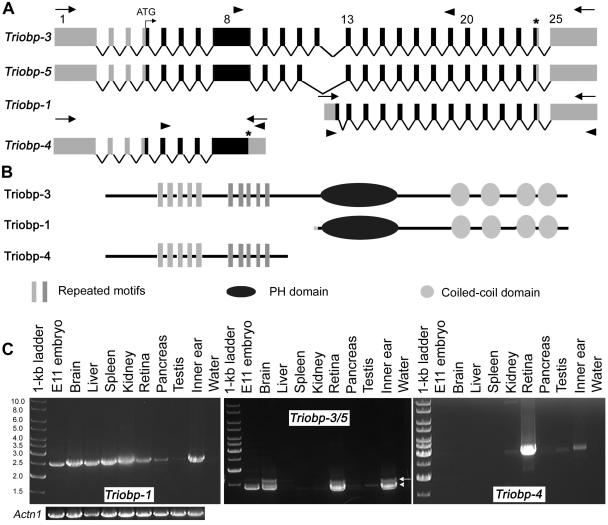
Genomic structure of Triobp mouse isoforms and their expression in various tissues. A, Triobp splice variants. There are at least five different splice variants present in the mouse inner ear, including the previously reported short isoforms (Triobp-1 and Triobp-2). Locations of the primers used for RT-PCR expression profile are shown as arrowheads. B, Predicted protein products of different Triobp splice variants. Triobp-4 does not encode the PH domain and the coiled-coil regions. Triobp-1 encodes only the PH domain and the four coiled-coil domains. C, Expression profiles of Triobp-1, Triobp-3/5, and Triobp-4. PCR primers (table 2), shown as arrowheads in panel A, were designed to amplify each isoform from cDNA prepared from different mouse tissues. Left profile, Triobp-1 is widely expressed, whereas other isoforms have a more limited pattern of expression. Triobp-3 (arrow) is present in the brain, liver (faint band), kidney (faint band), retina (P15), and inner ear (P1–P5). Middle profile,Triobp-5 (arrowhead) is found in mouse embryo cDNA (E11.5), brain, kidney (faint band), retina, pancreas (faint band), and inner ear. Right profile, Triobp-4 seems to be expressed in kidney (faint band), retina, testis (faint band), and inner ear. These transcripts do not provide an exhaustive list of the isoforms of Triobp. A smaller, uncharacterized cDNA is present in many tissues (middle profile). There are 33 inner-ear RIKEN ESTs for Triobp (168–488 bp; not shown). PCR amplification of Actn1 cDNA encoding α-actinin was used as a control for the quality and quantity of RT-PCR templates.
References
Web Resources
-
- Center for Medical Genetics, Marshfield Medical Research Foundation, http://research.marshfieldclinic.org/genetics/
-
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/ (for TRIOBP1 [accession number NM_007032], mRNAs for TRIOBP [accession numbers AB051449 and AK096634], TRIOBP-6 [accession number DQ228005], TRIOBP-3 [accession number DQ228003], TRIOBP-4 [accession number DQ228004], Triobp-3 [accession number DQ228000], Triobp-4 [accession number DQ228002], and Triobp-5 [accession number DQ228001])
-
- Hereditary Hearing Loss Homepage, http://webhost.ua.ac.be/hhh/
-
- Online Mendelian Inheritance of Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for WSIV)
References
-
- Ahmed ZM, Riazuddin S, Ahmad J, Bernstein SL, Guo Y, Sabar MF, Sieving P, Riazuddin S, Griffith AJ, Friedman TB, Belyantseva IA, Wilcox ER (2003) PCDH15 is expressed in the neurosensory epithelium of the eye and ear and mutant alleles are responsible for both USH1F and DFNB23. Hum Mol Genet 12:3215–322310.1093/hmg/ddg358 - DOI - PubMed
-
- Ahmed ZM, Smith TN, Riazuddin S, Makishima T, Ghosh M, Bokhari S, Menon PS, Deshmukh D, Griffith AJ, Riazuddin S, Friedman TB, Wilcox ER (2002) Nonsyndromic recessive deafness DFNB18 and Usher syndrome type IC are allelic mutations of USHIC. Hum Genet 110:527–53110.1007/s00439-002-0732-4 - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
