

Control of SRF binding to CArG box chromatin regulates smooth muscle gene expression in vivo
- PMID: 16395403
- PMCID: PMC1323266
- DOI: 10.1172/JCI26505
Control of SRF binding to CArG box chromatin regulates smooth muscle gene expression in vivo
Abstract
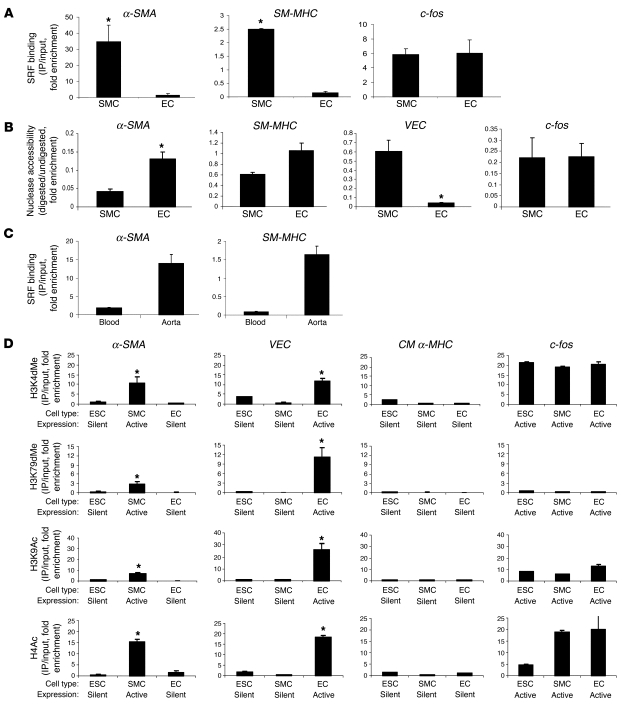
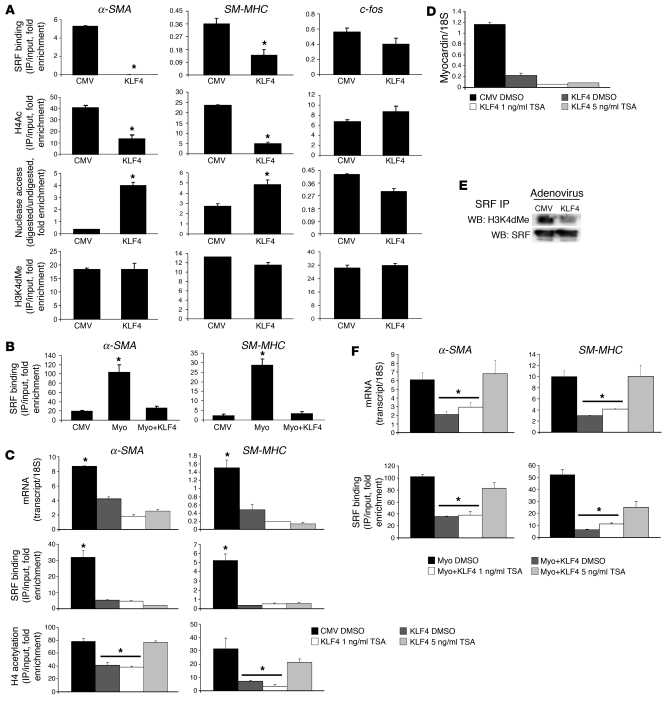
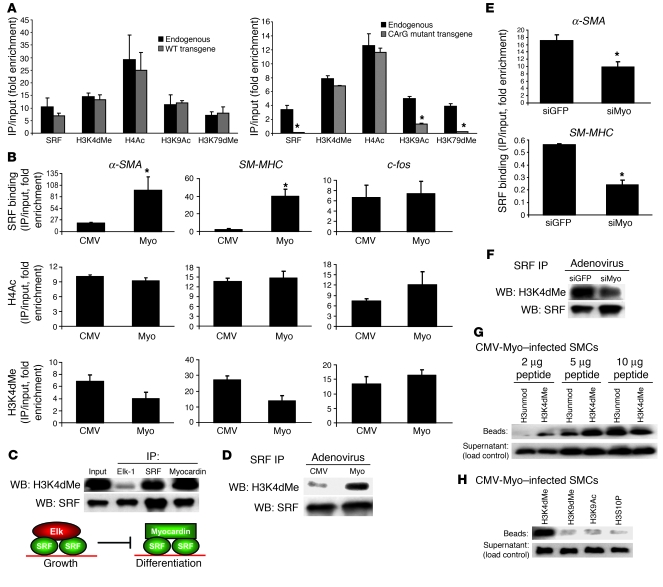
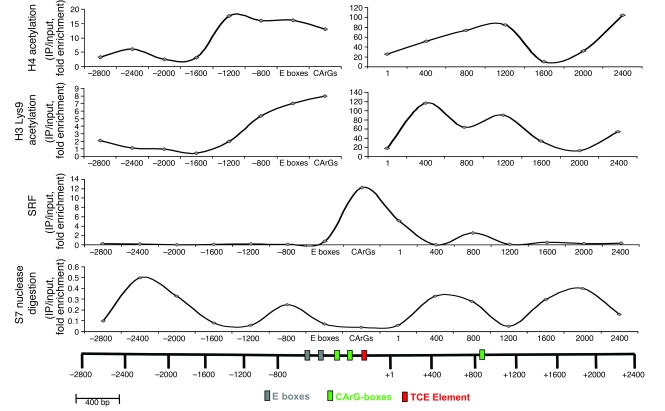
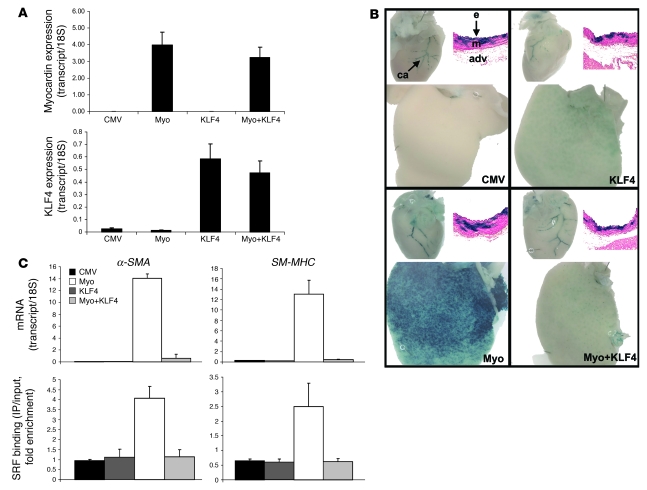
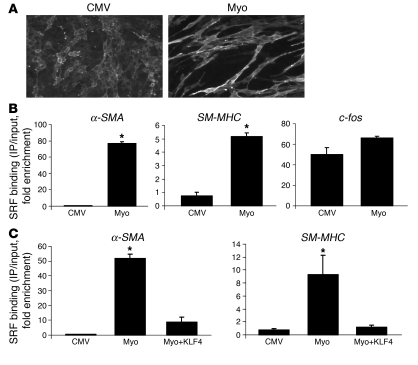
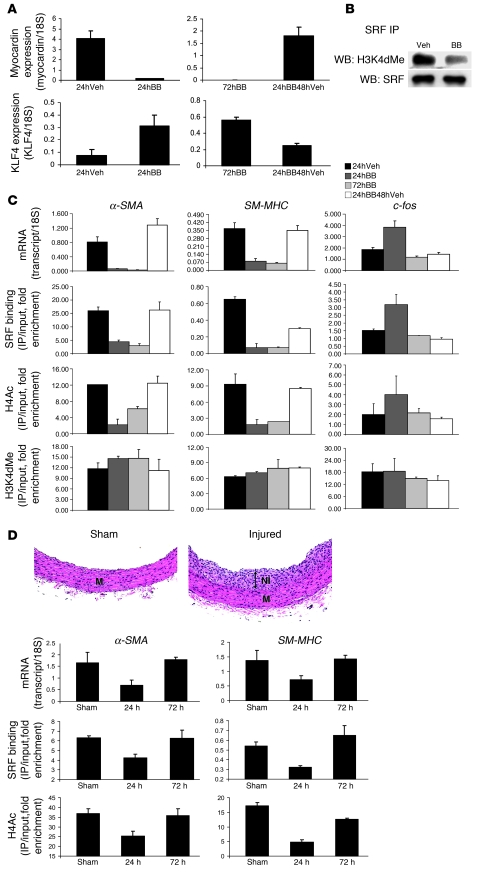
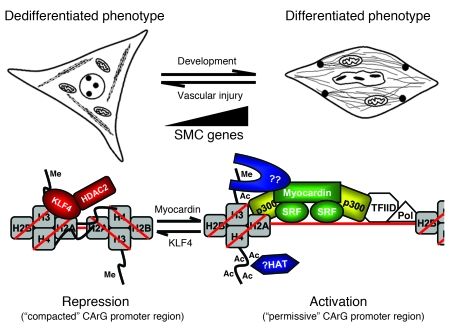
Precise control of SMC transcription plays a major role in vascular development and pathophysiology. Serum response factor (SRF) controls SMC gene transcription via binding to CArG box DNA sequences found within genes that exhibit SMC-restricted expression. However, the mechanisms that regulate SRF association with CArG box DNA within native chromatin of these genes are unknown. Here we report that SMC-restricted binding of SRF to murine SMC gene CArG box chromatin is associated with patterns of posttranslational histone modifications within this chromatin that are specific to the SMC lineage in culture and in vivo, including methylation and acetylation to histone H3 and H4 residues. We found that the promyogenic SRF coactivator myocardin increased SRF association with methylated histones and CArG box chromatin during activation of SMC gene expression. In contrast, the myogenic repressor Kruppel-like factor 4 recruited histone H4 deacetylase activity to SMC genes and blocked SRF association with methylated histones and CArG box chromatin during repression of SMC gene expression. Finally, we observed deacetylation of histone H4 coupled with loss of SRF binding during suppression of SMC differentiation in response to vascular injury. Taken together, these findings provide novel evidence that SMC-selective epigenetic control of SRF binding to chromatin plays a key role in regulation of SMC gene expression in response to pathophysiological stimuli in vivo.
Figures
References
-
- Owens G.K., Kumar M.S., Wamhoff B.R. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 2004;84:767–801. - PubMed
-
- Sata M., et al. Hematopoietic stem cells differentiate into vascular cells that participate in the pathogenesis of atherosclerosis. Nat. Med. 2002;8:403–409. - PubMed
-
- Glaser R., Lu M.M., Narula N., Epstein J.A. Smooth muscle cells, but not myocytes, of host origin in transplanted human hearts. Circulation. 2002;106:17–19. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
