

Supraspinal brain-derived neurotrophic factor signaling: a novel mechanism for descending pain facilitation
- PMID: 16399679
- PMCID: PMC6674294
- DOI: 10.1523/JNEUROSCI.3686-05.2006
Supraspinal brain-derived neurotrophic factor signaling: a novel mechanism for descending pain facilitation
Abstract
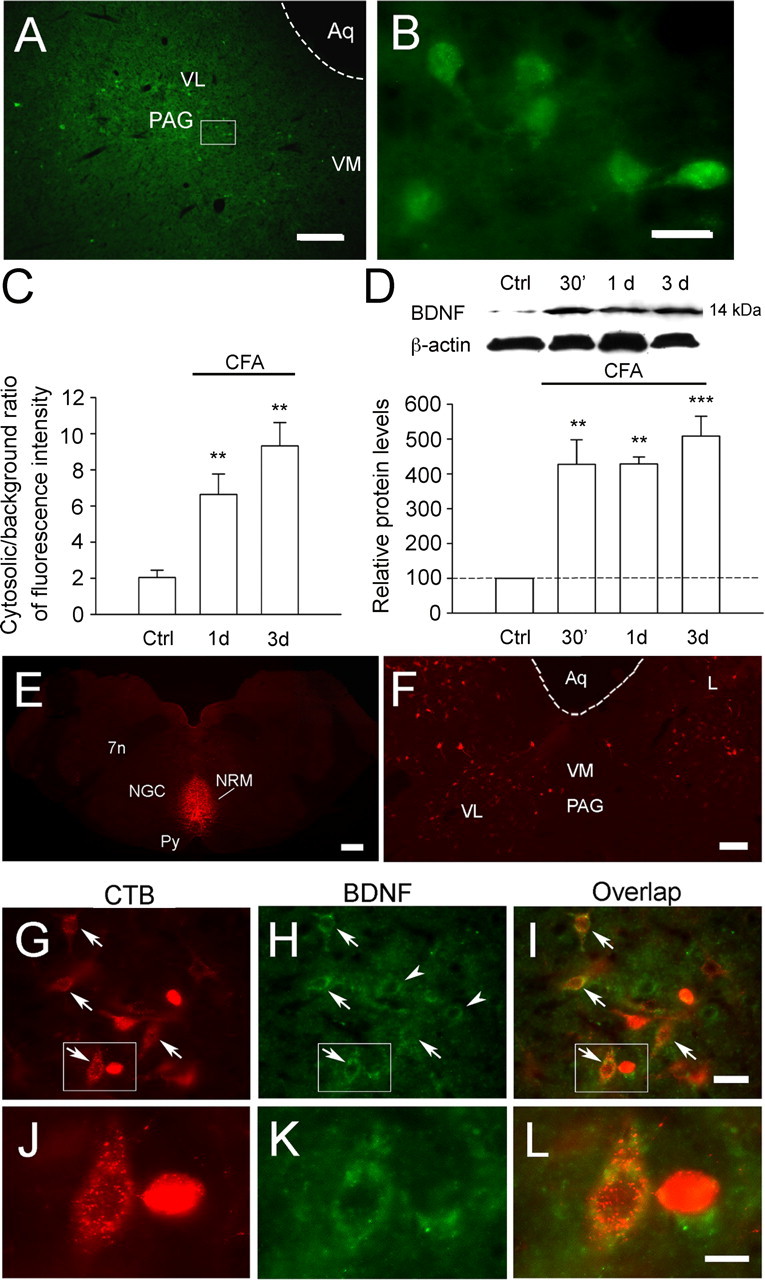
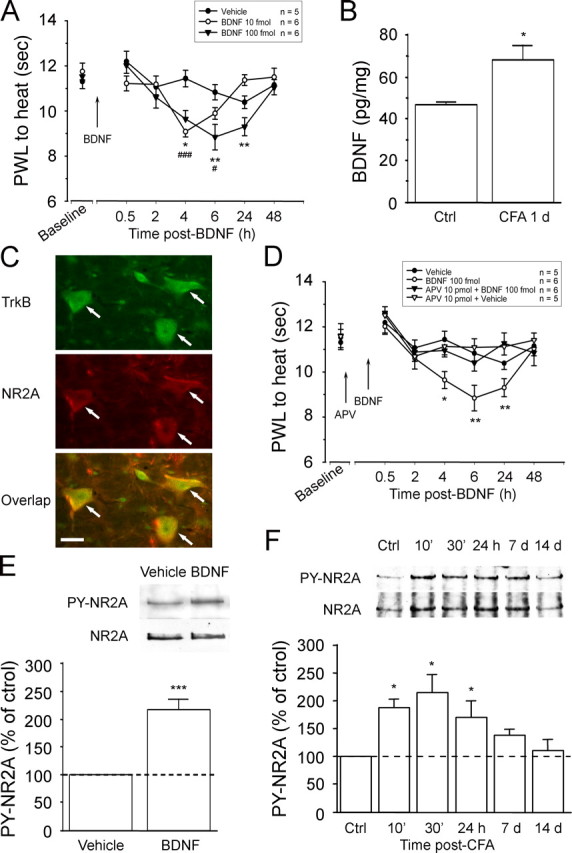
In the adult mammalian brain, brain-derived neurotrophic factor (BDNF) is critically involved in long-term synaptic plasticity. Here, we show that supraspinal BDNF-tyrosine kinase receptor B (TrkB) signaling contributes to pain facilitation. We show that BDNF-containing neurons in the periaqueductal gray (PAG), the central structure for pain modulation, project to and release BDNF in the rostral ventromedial medulla (RVM), a relay between the PAG and spinal cord. BDNF in PAG and TrkB phosphorylation in RVM neurons are upregulated after inflammation. Intra-RVM sequestration of BDNF and knockdown of TrkB by RNA interference attenuate inflammatory pain. Microinjection of BDNF (10-100 fmol) into the RVM facilitates nociception, which is dependent on NMDA receptors (NMDARs). In vitro studies with RVM slices show that BDNF induces tyrosine phosphorylation of the NMDAR NR2A subunit in RVM via a signal transduction cascade involving IP(3), PKC, and Src. The supraspinal BDNF-TrkB signaling represents a previously unknown mechanism underlying the development of persistent pain. Our findings also caution that application of BDNF for recovery from CNS disorders could lead to undesirable central pain.
Figures






References
-
- Akaneya Y, Jiang B, Tsumoto T (2005) RNAi-induced gene silencing by local electroporation in targeting brain region. J Neurophysiol 93: 594–602. - PubMed
-
- Cui M, Feng Y, McAdoo DJ, Willis WD (1999) Periaqueductal gray stimulation-induced inhibition of nociceptive dorsal horn neurons in rats is associated with the release of norepinephrine, serotonin, and amino acids. J Pharmacol Exp Ther 289: 868–876. - PubMed
-
- Di Luca M, Gardoni F, Finardi A, Pagliardini S, Cattabeni F, Battaglia G, Missale C (2001) NMDA receptor subunits are phosphorylated by activation of neurotrophin receptors in PSD of rat spinal cord. NeuroReport 12: 1301–1305. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous