

Using linkage genome scans to improve power of association in genome scans
- PMID: 16400608
- PMCID: PMC1380233
- DOI: 10.1086/500026
Using linkage genome scans to improve power of association in genome scans
Abstract
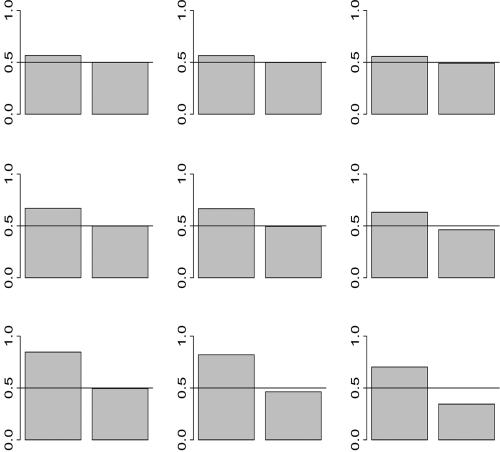
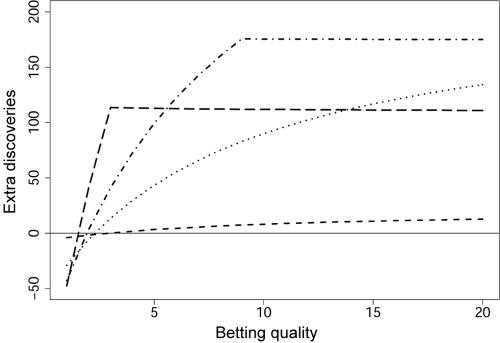
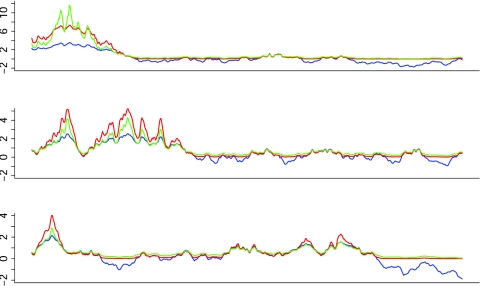
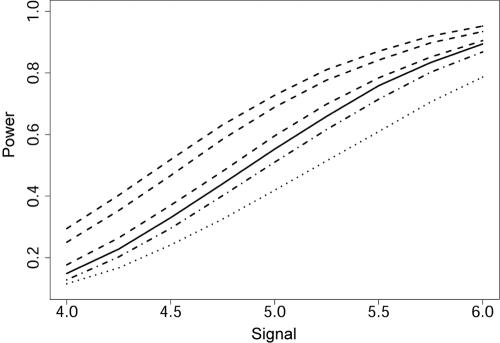
Scanning the genome for association between markers and complex diseases typically requires testing hundreds of thousands of genetic polymorphisms. Testing such a large number of hypotheses exacerbates the trade-off between power to detect meaningful associations and the chance of making false discoveries. Even before the full genome is scanned, investigators often favor certain regions on the basis of the results of prior investigations, such as previous linkage scans. The remaining regions of the genome are investigated simultaneously because genotyping is relatively inexpensive compared with the cost of recruiting participants for a genetic study and because prior evidence is rarely sufficient to rule out these regions as harboring genes with variation of conferring liability (liability genes). However, the multiple testing inherent in broad genomic searches diminishes power to detect association, even for genes falling in regions of the genome favored a priori. Multiple testing problems of this nature are well suited for application of the false-discovery rate (FDR) principle, which can improve power. To enhance power further, a new FDR approach is proposed that involves weighting the hypotheses on the basis of prior data. We present a method for using linkage data to weight the association P values. Our investigations reveal that if the linkage study is informative, the procedure improves power considerably. Remarkably, the loss in power is small, even when the linkage study is uninformative. For a class of genetic models, we calculate the sample size required to obtain useful prior information from a linkage study. This inquiry reveals that, among genetic models that are seemingly equal in genetic information, some are much more promising than others for this mode of analysis.
Figures
References
Web Resource
-
- University of Pittsburgh Medical Center, http://wpicr.wpic.pitt.edu/wpiccompgen/
References
-
- Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B 57:289–300
-
- ——— (1997) Multiple hypothesis testing with weights. Scand J Stat 24:407–418 10.1111/1467-9469.00072 - DOI
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
