

Spectrum of CHD7 mutations in 110 individuals with CHARGE syndrome and genotype-phenotype correlation
- PMID: 16400610
- PMCID: PMC1380237
- DOI: 10.1086/500273
Spectrum of CHD7 mutations in 110 individuals with CHARGE syndrome and genotype-phenotype correlation
Abstract
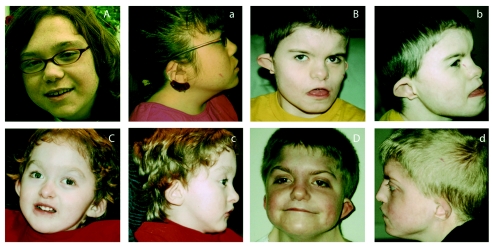
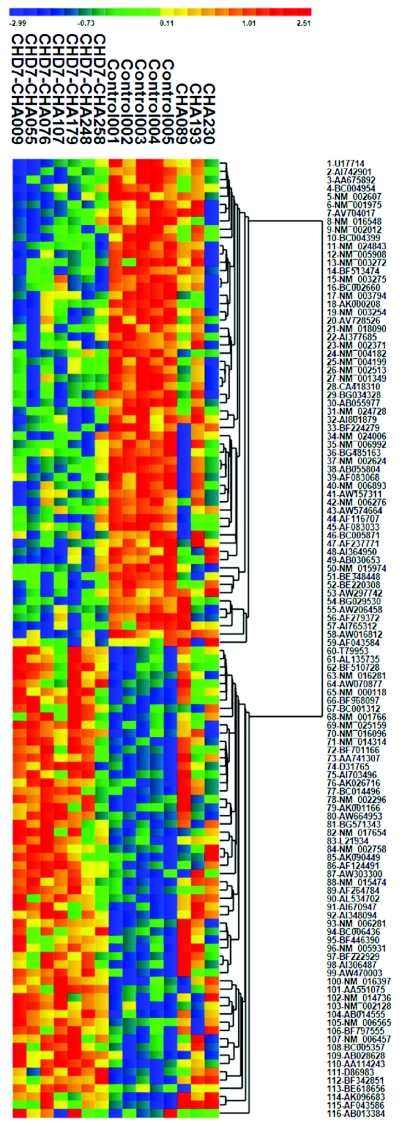
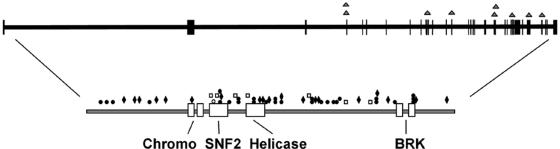
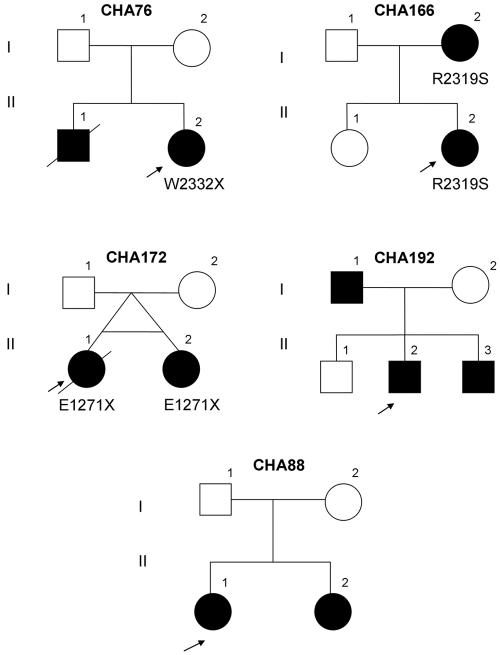
CHARGE syndrome is a well-established multiple-malformation syndrome with distinctive consensus diagnostic criteria. Characteristic associated anomalies include ocular coloboma, choanal atresia, cranial nerve defects, distinctive external and inner ear abnormalities, hearing loss, cardiovascular malformations, urogenital anomalies, and growth retardation. Recently, mutations of the chromodomain helicase DNA-binding protein gene CHD7 were reported to be a major cause of CHARGE syndrome. We sequenced the CHD7 gene in 110 individuals who had received the clinical diagnosis of CHARGE syndrome, and we detected mutations in 64 (58%). Mutations were distributed throughout the coding exons and conserved splice sites of CHD7. Of the 64 mutations, 47 (73%) predicted premature truncation of the protein. These included nonsense and frameshift mutations, which most likely lead to haploinsufficiency. Phenotypically, the mutation-positive group was more likely to exhibit cardiovascular malformations (54 of 59 in the mutation-positive group vs. 30 of 42 in the mutation-negative group; P=.014), coloboma of the eye (55 of 62 in the mutation-positive group vs. 30 of 43 in the mutation-negative group; P=.022), and facial asymmetry, often caused by seventh cranial nerve abnormalities (36 of 56 in the mutation-positive group vs. 13 of 39 in the mutation-negative group; P=.004). Mouse embryo whole-mount and section in situ hybridization showed the expression of Chd7 in the outflow tract of the heart, optic vesicle, facio-acoustic preganglion complex, brain, olfactory pit, and mandibular component of the first branchial arch. Microarray gene-expression analysis showed a signature pattern of gene-expression differences that distinguished the individuals with CHARGE syndrome with CHD7 mutation from the controls. We conclude that cardiovascular malformations, coloboma, and facial asymmetry are common findings in CHARGE syndrome caused by CHD7 mutation.
Figures

References
Web Resources
-
- Baylor College of Medicine Cardiovascular Genetics, http://www.cardiogene.org/
-
- ChipST2C, http://chipst2c.org/ChipST2C.html
-
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/ (for gene sequences [accession number NM_017780], clones RP11-33I11 [accession number AC113143] and RP11-174G1 [accession number AC022182], and cDNA fragment of KIAA1416 [accession number BC034239])
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for CHARGE syndrome)
References
-
- Amiel J, Attiee-Bitach T, Marianowski R, Cormier-Daire V, Abadie V, Bonnet D, Gonzales M, Chemouny S, Brunelle F, Munnich A, Manach Y, Lyonnet S (2001) Temporal bone anomaly proposed as a major criteria for diagnosis of CHARGE syndrome. Am J Med Genet 99:124–12710.1002/1096-8628(20010301)99:2<124::AID-AJMG1114>3.0.CO;2-9 - DOI - PubMed
-
- Blake KD, Davenport SL, Hall BD, Hefner MA, Pagon RA, Williams MS, Lin AE, Graham JM Jr (1998) CHARGE association: an update and review for the primary pediatrician. Clin Pediatr (Phila) 37:159–173 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
