

Mutations in the translated region of the lactase gene (LCT) underlie congenital lactase deficiency
- PMID: 16400612
- PMCID: PMC1380240
- DOI: 10.1086/500053
Mutations in the translated region of the lactase gene (LCT) underlie congenital lactase deficiency
Abstract
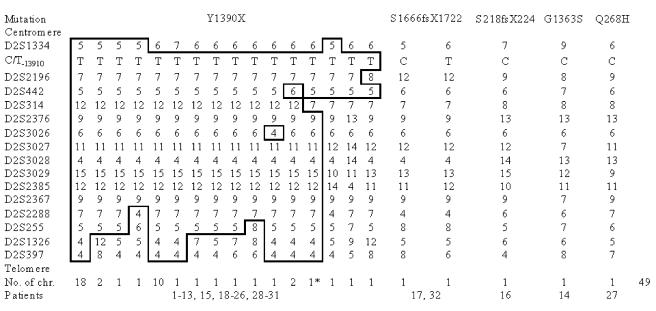
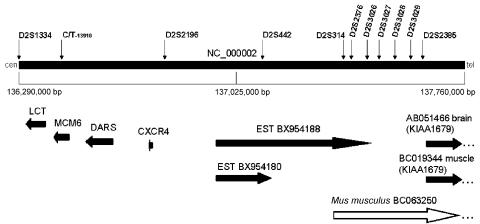
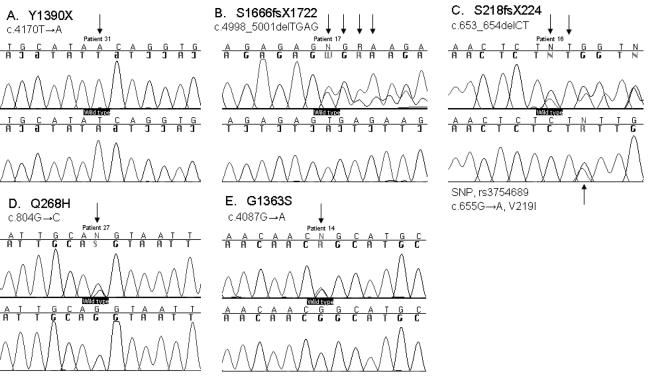
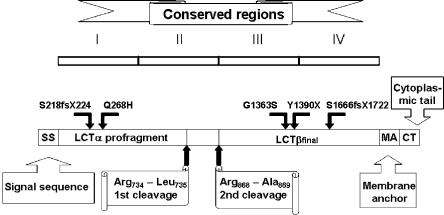
Congenital lactase deficiency (CLD) is a severe gastrointestinal disorder characterized by watery diarrhea in infants fed with breast milk or other lactose-containing formulas. We initially assigned the CLD locus by linkage and linkage disequilibrium on 2q21 in 19 Finnish families. Here we report the molecular background of CLD via characterization of five distinct mutations in the coding region of the lactase (LCT) gene. Twenty-seven patients out of 32 (84%) were homozygous for a nonsense mutation, c.4170T-->A (Y1390X), designated "Fin(major)." Four rare mutations--two that result in a predicted frameshift and early truncation at S1666fsX1722 and S218fsX224 and two point mutations that result in substitutions Q268H and G1363S of the 1,927-aa polypeptide--confirmed the lactase mutations as causative for CLD. These findings facilitate genetic testing in clinical practice and enable genetic counseling for this severe disease. Further, our data demonstrate that, in contrast to common adult-type hypolactasia (lactose intolerance) caused by a variant of the regulatory element, the severe infancy form represents the outcome of mutations affecting the structure of the protein inactivating the enzyme.
Figures
References
Web Resources
-
- Baylor College of Medicine (BCM) Search Launcher, http://searchlauncher.bcm.tmc.edu/
-
- Marshfield Clinic Research Foundation Genetic Map, http://research.marshfieldclinic.org/
-
- National Center for Biotechnology Information BLAST, http://www.ncbi.nlm.nih.gov/BLAST/
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim
-
- UCSC Genome Browser, http://genome.ucsc.edu/
References
-
- Freiburghaus AU, Schmitz J, Schindler M, Rotthauwe HW, Kuitunen P, Launiala K, Hadorn B (1976) Protein patterns of brush-border fragments in congenital lactose malabsorption and in specific hypolactasia of the adult. N Engl J Med 294:1030–1032 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
