

A mutation in para-hydroxybenzoate-polyprenyl transferase (COQ2) causes primary coenzyme Q10 deficiency
- PMID: 16400613
- PMCID: PMC1380241
- DOI: 10.1086/500092
A mutation in para-hydroxybenzoate-polyprenyl transferase (COQ2) causes primary coenzyme Q10 deficiency
Abstract
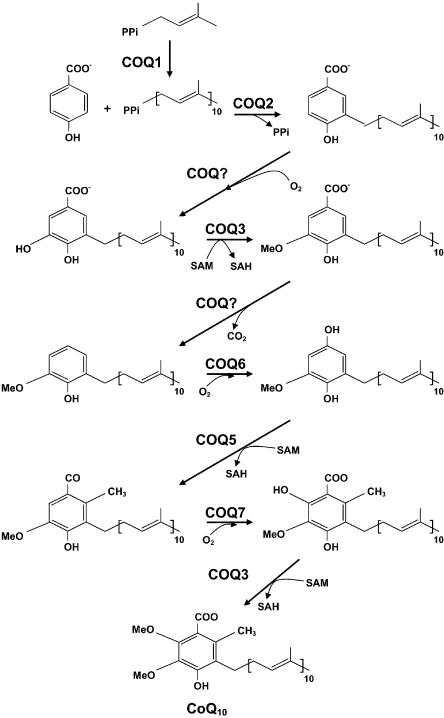
Ubiquinone (coenzyme Q(10) or CoQ(10)) is a lipid-soluble component of virtually all cell membranes, where it functions as a mobile electron and proton carrier. CoQ(10) deficiency is inherited as an autosomal recessive trait and has been associated with three main clinical phenotypes: a predominantly myopathic form with central nervous system involvement, an infantile encephalomyopathy with renal dysfunction, and an ataxic form with cerebellar atrophy. In two siblings of consanguineous parents with the infantile form of CoQ(10) deficiency, we identified a homozygous missense mutation in the COQ2 gene, which encodes para-hydroxybenzoate-polyprenyl transferase. The A-->G transition at nucleotide 890 changes a highly conserved tyrosine to cysteine at amino acid 297 within a predicted transmembrane domain. Radioisotope assays confirmed a severe defect of CoQ(10) biosynthesis in the fibroblasts of one patient. This mutation in COQ2 is the first molecular cause of primary CoQ(10) deficiency.
Figures

References
Web Resources
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for CoQ10 deficiency)
References
-
- Aure K, Benoist JF, Ogier de Baulny H, Romero NB, Rigal O, Lombes A (2004) Progression despite replacement of a myopathic form of coenzyme Q10 defect. Neurology 63:727–729 - PubMed
-
- Brea-Calvo G, Rodriguez-Hernandez A, Fernandez-Ayala DJM, Navas P, Sanchez-Alcazar JA. Chemotherapy induces an increase in coenzyme Q10 levels in cancer cell lines. Free Radic Biol Med (in press) - PubMed
-
- Date H, Onodera O, Tanaka H, Iwabuchi K, Uekawa K, Igarashi S, Koike R, Hiroi T, Yuasa T, Awaya Y, Sakai T, Takahashi T, Nagatomo H, Sekijima Y, Kawachi I, Takiyama Y, Nishizawa M, Fukuhara N, Saito K, Sugano S, Tsuji S (2001) Early-onset ataxia with ocular motor apraxia and hypoalbuminemia is caused by mutations in a new HIT superfamily gene. Nat Genet 29:184–18810.1038/ng1001-184 - DOI - PubMed
-
- Di Giovanni S, Mirabella M, Spinazzola A, Crociani P, Silvestri G, Broccolini A, Tonali P, Di Mauro S, Servidei S (2001) Coenzyme Q10 reverses pathological phenotype and reduces apoptosis in familial CoQ10 deficiency. Neurology 57:515–518 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
