

Compound developmental eye disorders following inactivation of TGFbeta signaling in neural-crest stem cells
- PMID: 16403239
- PMCID: PMC1414066
- DOI: 10.1186/jbiol29
Compound developmental eye disorders following inactivation of TGFbeta signaling in neural-crest stem cells
Abstract
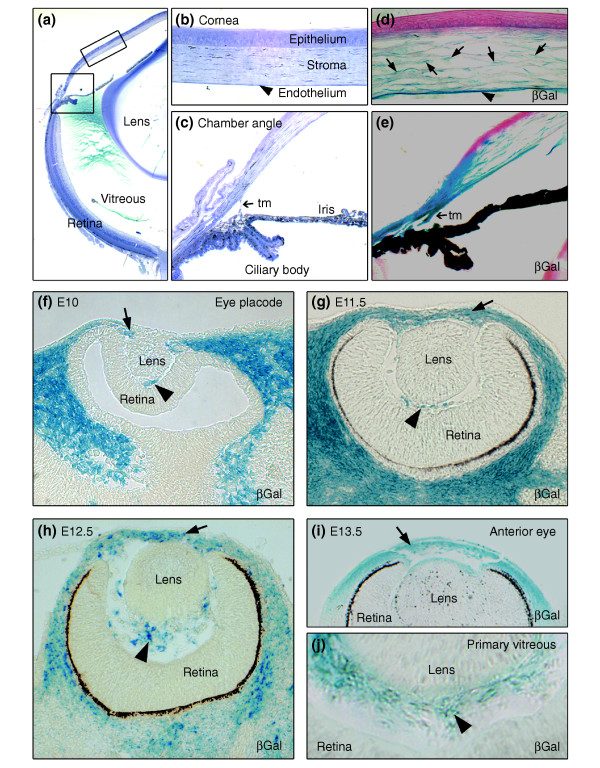
Background: Development of the eye depends partly on the periocular mesenchyme derived from the neural crest (NC), but the fate of NC cells in mammalian eye development and the signals coordinating the formation of ocular structures are poorly understood.
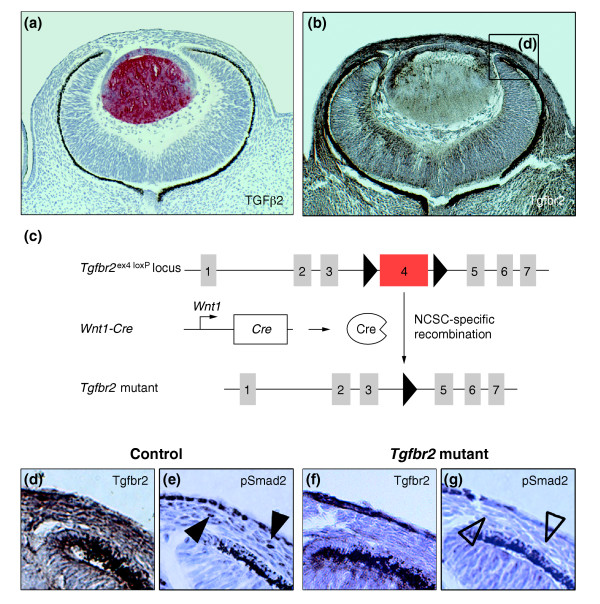
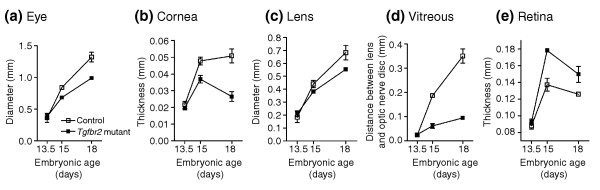
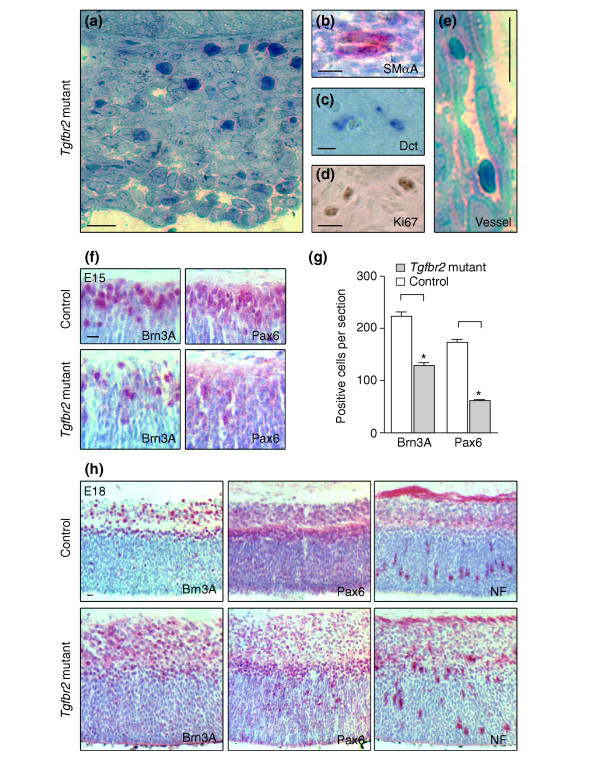
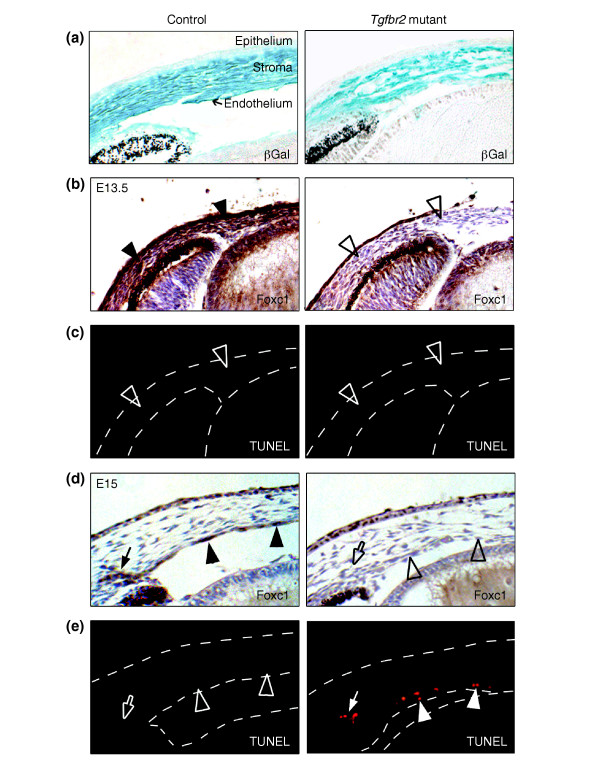
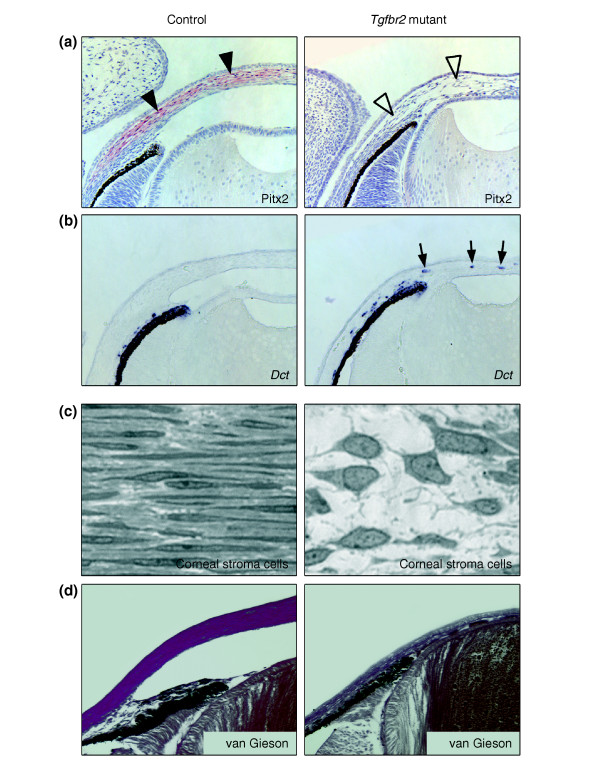
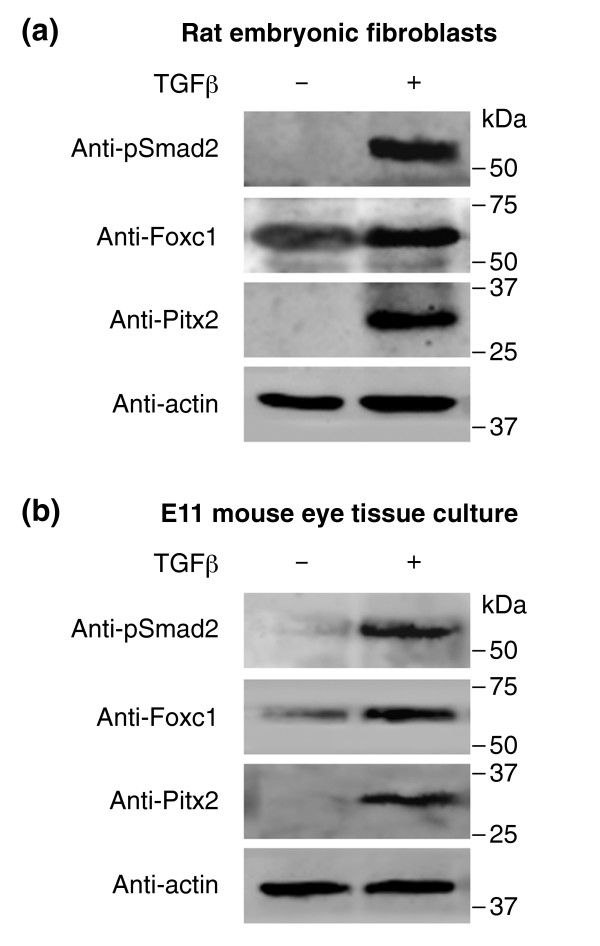
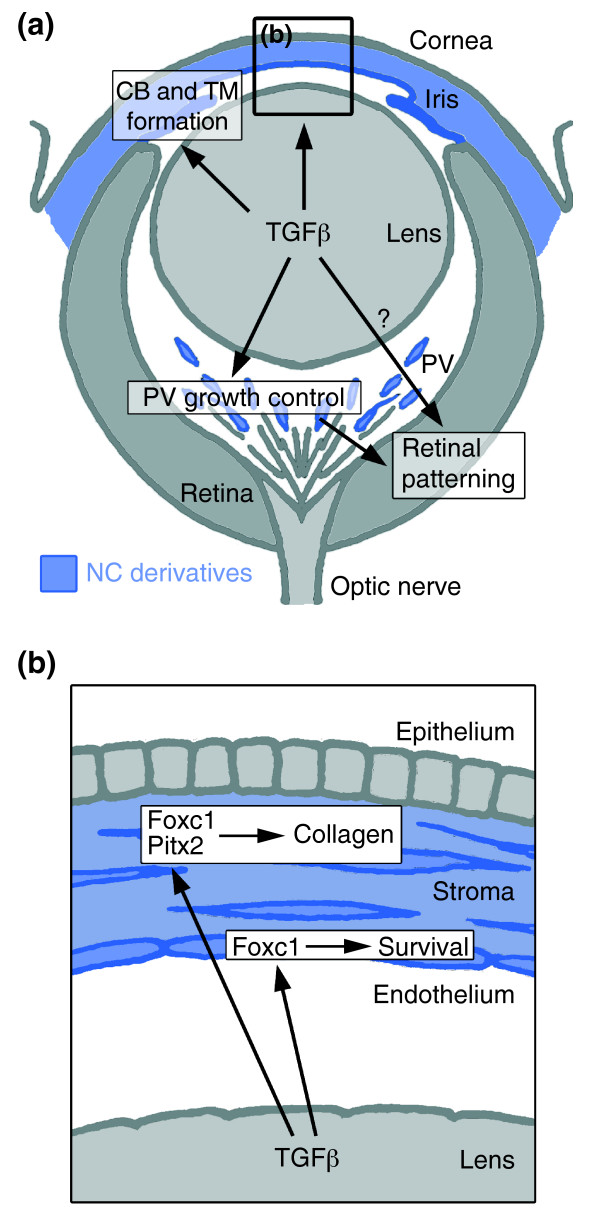
Results: Here we reveal distinct NC contributions to both anterior and posterior mesenchymal eye structures and show that TGFbeta signaling in these cells is crucial for normal eye development. In the anterior eye, TGFbeta2 released from the lens is required for the expression of transcription factors Pitx2 and Foxc1 in the NC-derived cornea and in the chamber-angle structures of the eye that control intraocular pressure. TGFbeta enhances Foxc1 and induces Pitx2 expression in cell cultures. As in patients carrying mutations in PITX2 and FOXC1, TGFbeta signal inactivation in NC cells leads to ocular defects characteristic of the human disorder Axenfeld-Rieger's anomaly. In the posterior eye, NC cell-specific inactivation of TGFbeta signaling results in a condition reminiscent of the human disorder persistent hyperplastic primary vitreous. As a secondary effect, retinal patterning is also disturbed in mutant mice.
Conclusion: In the developing eye the lens acts as a TGFbeta signaling center that controls the development of eye structures derived from the NC. Defective TGFbeta signal transduction interferes with NC-cell differentiation and survival anterior to the lens and with normal tissue morphogenesis and patterning posterior to the lens. The similarity to developmental eye disorders in humans suggests that defective TGFbeta signal modulation in ocular NC derivatives contributes to the pathophysiology of these diseases.
Figures

References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
