

Nonsteroidal anti-inflammatory drugs repress beta-secretase gene promoter activity by the activation of PPARgamma
- PMID: 16407166
- PMCID: PMC1326151
- DOI: 10.1073/pnas.0503839103
Nonsteroidal anti-inflammatory drugs repress beta-secretase gene promoter activity by the activation of PPARgamma
Abstract
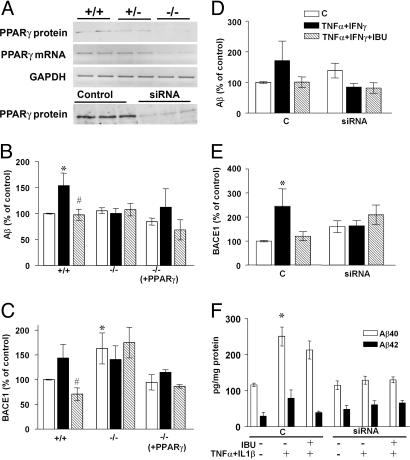
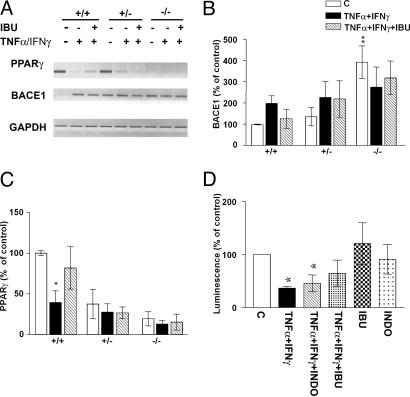
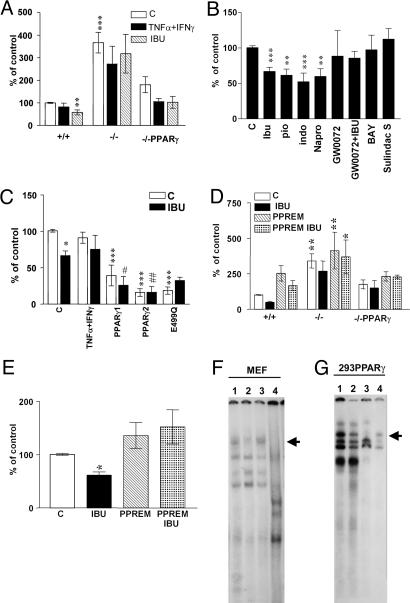
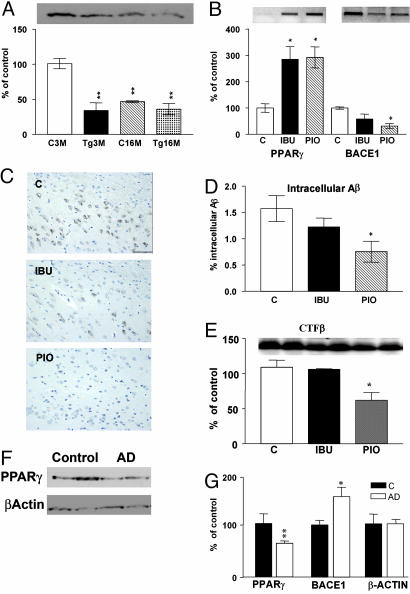
Epidemiological evidence suggests that nonsteroidal anti-inflammatory drugs (NSAIDs) decrease the risk for Alzheimer's disease (AD). Certain NSAIDs can activate the peroxisome proliferator-activated receptor-gamma (PPARgamma), which is a nuclear transcriptional regulator. Here we show that PPARgamma depletion potentiates beta-secretase [beta-site amyloid precursor protein cleaving enzyme (BACE1)] mRNA levels by increasing BACE1 gene promoter activity. Conversely, overexpression of PPARgamma, as well as NSAIDs and PPARgamma activators, reduced BACE1 gene promoter activity. These results suggested that PPARgamma could be a repressor of BACE1. We then identified a PPARgamma responsive element (PPRE) in the BACE1 gene promoter. Mutagenesis of the PPRE abolished the binding of PPARgamma to the PPRE and increased BACE1 gene promoter activity. Furthermore, proinflammatory cytokines decreased PPARgamma gene transcription, and this effect was supressed by NSAIDs. We also demonstrate that in vivo treatment with PPARgamma agonists increased PPARgamma and reduced BACE1 mRNA and intracellular beta-amyloid levels. Interestingly, brain extracts from AD patients showed decreased PPARgamma expression and binding to PPRE in the BACE1 gene promoter. Our data strongly support a major role of PPARgamma in the modulation of amyloid-beta generation by inflammation and suggest that the protective mechanism of NSAIDs in AD involves activation of PPARgamma and decreased BACE1 gene transcription.
Figures
References
-
- Vassar, R., Bennett, B. D., Babu-Khan, S., Kahn, S., Mendiaz, E. A., Denis, P., Teplow, D. B., Ross, S, Amarante, P., Loeloff, R., et al. (1999) Science 286, 735-741. - PubMed
-
- Luo, Y., Bolon, B., Damore, M. A., Fitzpatrick, D., Liu, H., Zhang, J., Yan, Q., Vassar, R. & Citron, M. (2003) Neurobiol. Dis. 14, 81-88. - PubMed
-
- Ohno, M., Sametsky, E. A., Younkin, L. H., Oakley, H., Younkin, S. G., Citron, M., Vassar, R. & Disterhoft, J. F. (2004) Neuron 41, 27-33. - PubMed
-
- Tamagno, E., Bardini, P., Obbili, A., Vitali, A., Borghi, R., Zaccheo, D., Pronzato, M. A., Danni, O., Smith, M. A., Perry, G. & Tabato, M. (2002) Neurobiol. Dis. 10, 279-288. - PubMed
-
- Hartlage-Rubsamen, M., Zeitschel, U., Apelt, J., Gartner, U., Franke, H., Stahl, T., Gunther, A., Schliebs, R., Penkowa, M., Bigl, V. & Rossner, S. (2003) Glia 4, 169-179. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
