

MaGe: a microbial genome annotation system supported by synteny results
- PMID: 16407324
- PMCID: PMC1326237
- DOI: 10.1093/nar/gkj406
MaGe: a microbial genome annotation system supported by synteny results
Abstract
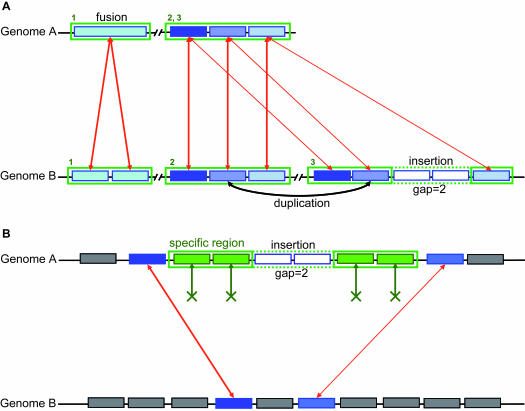
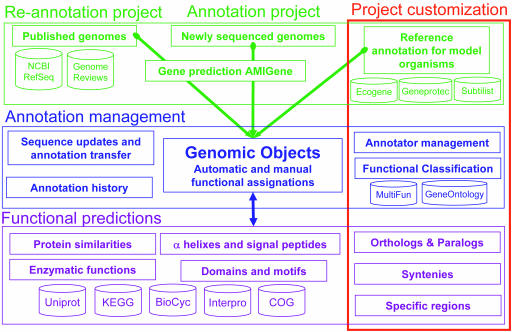
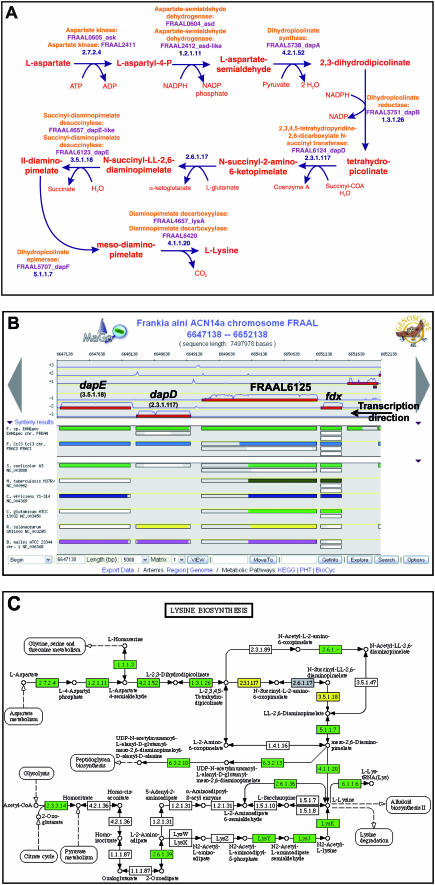
Magnifying Genomes (MaGe) is a microbial genome annotation system based on a relational database containing information on bacterial genomes, as well as a web interface to achieve genome annotation projects. Our system allows one to initiate the annotation of a genome at the early stage of the finishing phase. MaGe's main features are (i) integration of annotation data from bacterial genomes enhanced by a gene coding re-annotation process using accurate gene models, (ii) integration of results obtained with a wide range of bioinformatics methods, among which exploration of gene context by searching for conserved synteny and reconstruction of metabolic pathways, (iii) an advanced web interface allowing multiple users to refine the automatic assignment of gene product functions. MaGe is also linked to numerous well-known biological databases and systems. Our system has been thoroughly tested during the annotation of complete bacterial genomes (Acinetobacter baylyi ADP1, Pseudoalteromonas haloplanktis, Frankia alni) and is currently used in the context of several new microbial genome annotation projects. In addition, MaGe allows for annotation curation and exploration of already published genomes from various genera (e.g. Yersinia, Bacillus and Neisseria). MaGe can be accessed at http://www.genoscope.cns.fr/agc/mage.
Figures

References
-
- Galperin M.Y., Koonin E.V. Sources of systematic error in functional annotation of genomes: domain rearrangement, non-orthologous gene displacement and operon disruption. In Silico Biol. 1998;1:55–67. - PubMed
-
- Brenner S.E. Errors in genome annotation. Trends Genet. 1999;15:132–133. - PubMed
-
- Gaasterland T., Sensen C.W. MAGPIE: automated genome interpretation. Trends Genet. 1996;12:76–78. - PubMed
