

N-(4-hydroxyphenyl)retinamide-induced apoptosis triggered by reactive oxygen species is mediated by activation of MAPKs in head and neck squamous carcinoma cells
- PMID: 16407847
- PMCID: PMC1458365
- DOI: 10.1038/sj.onc.1209303
N-(4-hydroxyphenyl)retinamide-induced apoptosis triggered by reactive oxygen species is mediated by activation of MAPKs in head and neck squamous carcinoma cells
Abstract
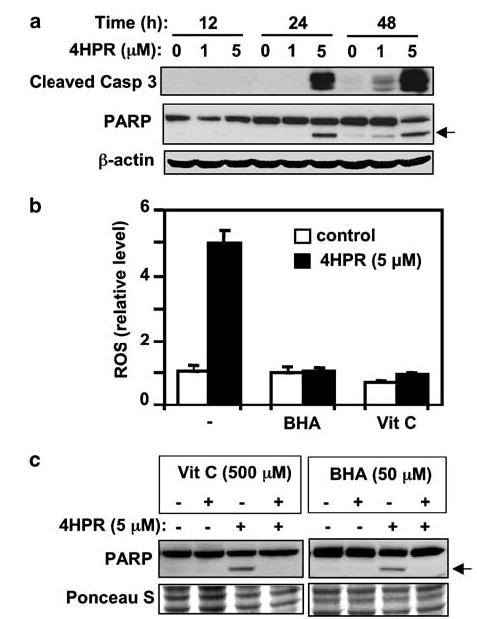
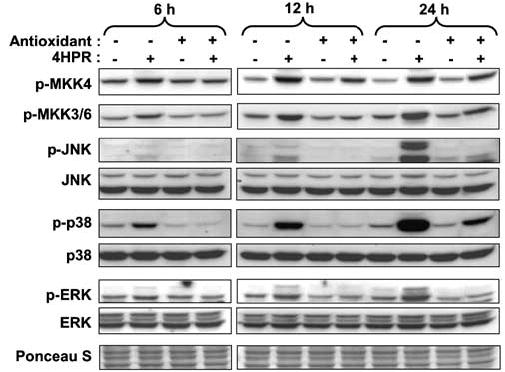
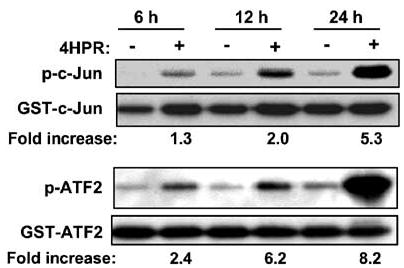
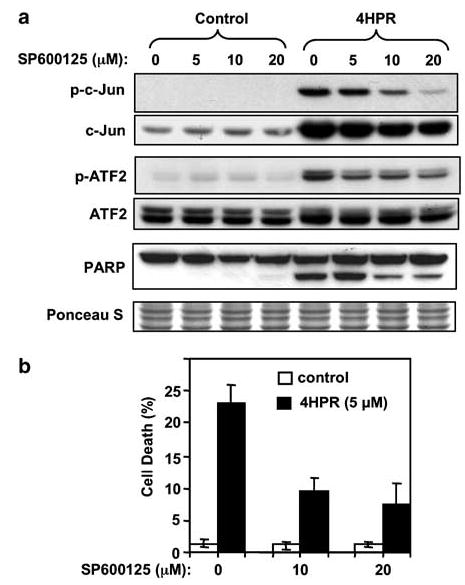
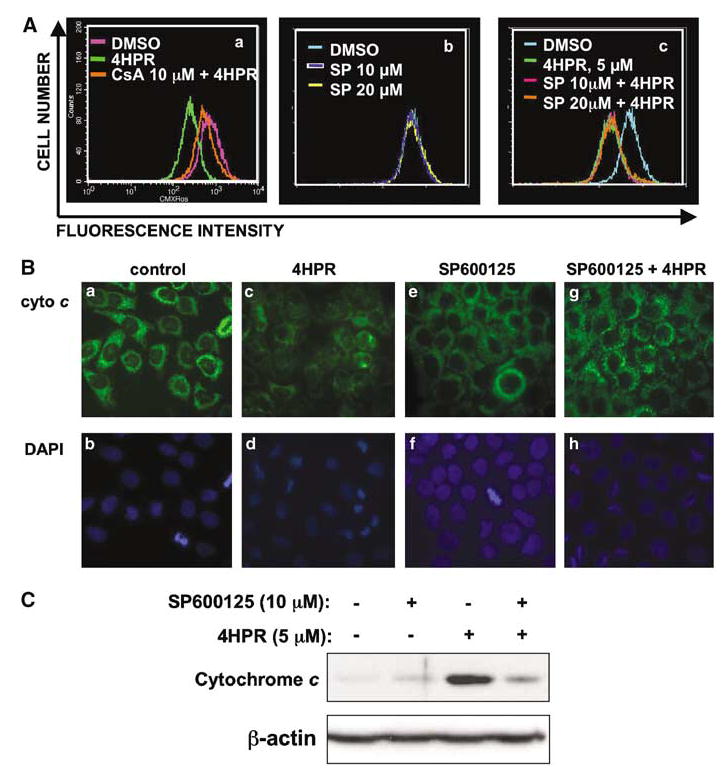
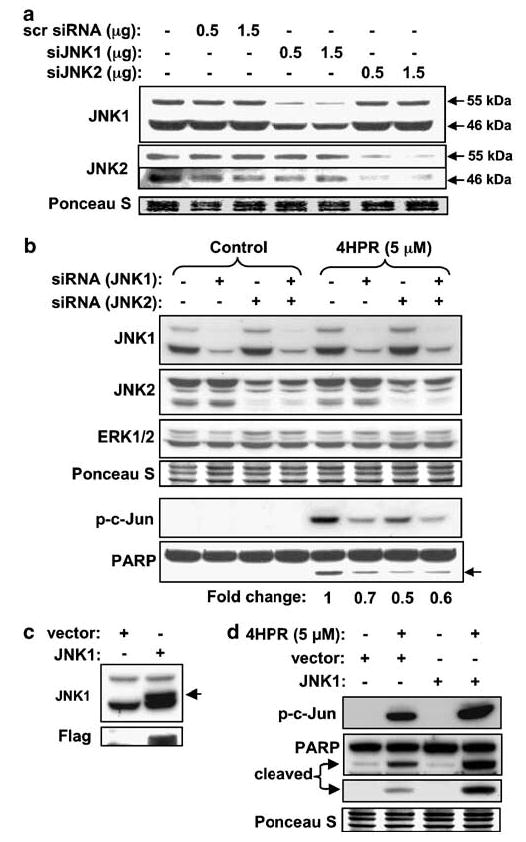
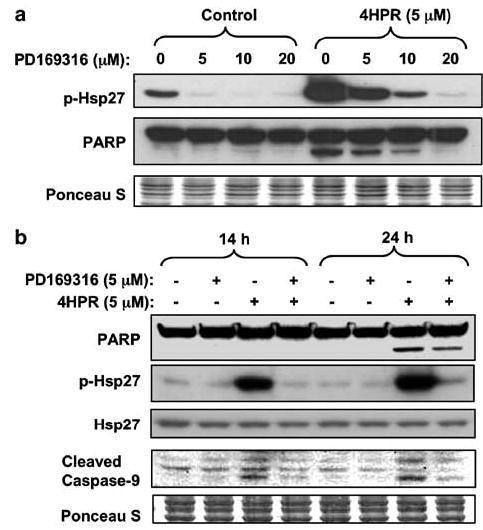
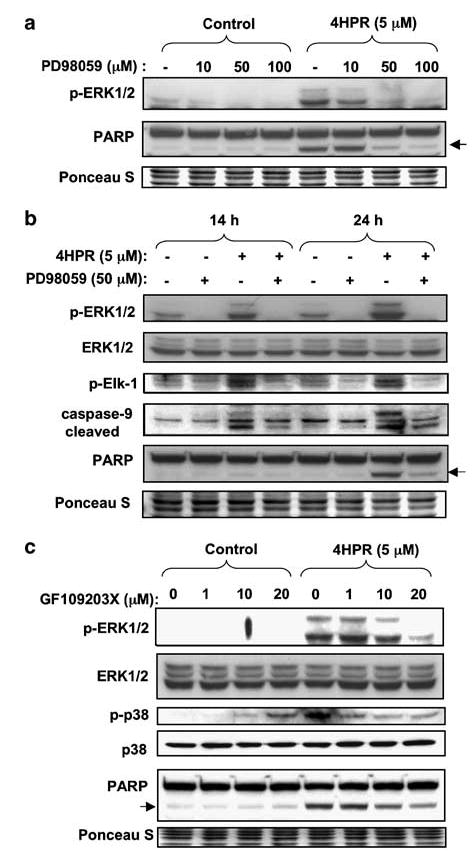
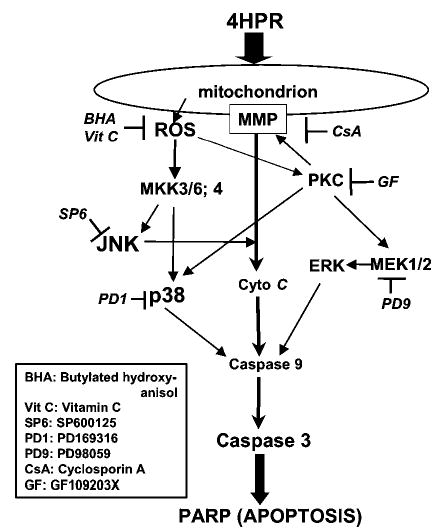
N-(4-hydroxyphenyl)retinamide (4HPR), a synthetic retinoid effective in cancer chemoprevention and therapy, is thought to act via apoptosis induction resulting from increased reactive oxygen species (ROS) generation. As ROS can activate MAP kinases and protein kinase C (PKC), we examined the role of such enzymes in 4HPR-induced apoptosis in HNSCC UMSCC22B cells. 4HPR increased ROS level within 1 h and induced activation of caspase 3 and PARP cleavage within 24 h. Activation of MKK3/6 and MKK4, JNK, p38 and ERK was detected between 6 and 12 h, increased up to 24 h and preceded apoptosis. 4HPR-induced activation of these kinases was abrogated by the antioxidants BHA and vitamin C. SP600125, a JNK inhibitor, suppressed 4HPR-induced c-Jun phosphorylation, cytochrome c release from mitochondria and apoptosis. Suppression of JNK1 and JNK2 using siRNA decreased, whereas overexpression of wild type-JNK1 enhanced 4HPR-induced apoptosis. PD169316, a p38, inhibitor suppressed phosphorylation of Hsp27 and apoptosis. PD98059, an MEK1/2 inhibitor, also suppressed ERK1/2 activation and apoptosis induced by 4HPR. Likewise, PKC inhibitor GF109203X suppressed ERK and p38 phosphorylation and PARP cleavage. These data indicate that 4HPR-induced apoptosis is triggered by ROS increase, leading to the activation of the mitogen-activated protein serine/threonine kinases JNK, p38, PKC and ERK, and subsequent apoptosis.
Figures
Similar articles
-
Induction of endoplasmic reticulum stress by the pro-apoptotic retinoid N-(4-hydroxyphenyl)retinamide via a reactive oxygen species-dependent mechanism in human head and neck cancer cells.Cancer Biol Ther. 2007 May;6(5):705-11. doi: 10.4161/cbt.6.5.3963. Epub 2007 Feb 3. Cancer Biol Ther. 2007. PMID: 17404501
-
Implication of mitochondria-derived reactive oxygen species, cytochrome C and caspase-3 in N-(4-hydroxyphenyl)retinamide-induced apoptosis in cervical carcinoma cells.Oncogene. 1999 Nov 4;18(46):6380-7. doi: 10.1038/sj.onc.1203024. Oncogene. 1999. PMID: 10597238
-
Increased level of the p67phox subunit of NADPH oxidase by 4HPR in head and neck squamous carcinoma cells.Int J Oncol. 2005 Sep;27(3):787-90. Int J Oncol. 2005. PMID: 16077929
-
Overexpression of extracellular-signal regulated kinases on oral squamous cell carcinoma.Oral Oncol. 2002 Jul;38(5):468-74. doi: 10.1016/s1368-8375(01)00104-x. Oral Oncol. 2002. PMID: 12110341 Review.
-
Mechanism of fenretinide (4-HPR)-induced cell death.Apoptosis. 2001 Oct;6(5):377-88. doi: 10.1023/a:1011342220621. Apoptosis. 2001. PMID: 11483862 Review.
Cited by
-
Cell signaling through protein kinase C oxidation and activation.Int J Mol Sci. 2012;13(9):10697-10721. doi: 10.3390/ijms130910697. Epub 2012 Aug 24. Int J Mol Sci. 2012. PMID: 23109817 Free PMC article. Review.
-
Apoptosis of bone marrow mesenchymal stem cells caused by homocysteine via activating JNK signal.PLoS One. 2013 May 7;8(5):e63561. doi: 10.1371/journal.pone.0063561. Print 2013. PLoS One. 2013. PMID: 23667638 Free PMC article.
-
Fenretinide (4-HPR) Targets Caspase-9, ERK 1/2 and the Wnt3a/β-Catenin Pathway in Medulloblastoma Cells and Medulloblastoma Cell Spheroids.PLoS One. 2016 Jul 1;11(7):e0154111. doi: 10.1371/journal.pone.0154111. eCollection 2016. PLoS One. 2016. PMID: 27367907 Free PMC article.
-
Cell Cycle Arrest and Apoptosis Induced by Porphyromonas gingivalis Require Jun N-Terminal Protein Kinase- and p53-Mediated p38 Activation in Human Trophoblasts.Infect Immun. 2018 Mar 22;86(4):e00923-17. doi: 10.1128/IAI.00923-17. Print 2018 Apr. Infect Immun. 2018. PMID: 29339463 Free PMC article.
-
Role for PKC δ in Fenretinide-Mediated Apoptosis in Lymphoid Leukemia Cells.J Signal Transduct. 2010 Jan 1;2010:584657. doi: 10.1155/2010/584657. J Signal Transduct. 2010. PMID: 20844597 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
