

Analysis of the human cytomegalovirus genomic region from UL146 through UL147A reveals sequence hypervariability, genotypic stability, and overlapping transcripts
- PMID: 16409621
- PMCID: PMC1360065
- DOI: 10.1186/1743-422X-3-4
Analysis of the human cytomegalovirus genomic region from UL146 through UL147A reveals sequence hypervariability, genotypic stability, and overlapping transcripts
Abstract
Background: Although the sequence of the human cytomegalovirus (HCMV) genome is generally conserved among unrelated clinical strains, some open reading frames (ORFs) are highly variable. UL146 and UL147, which encode CXC chemokine homologues are among these variable ORFs.
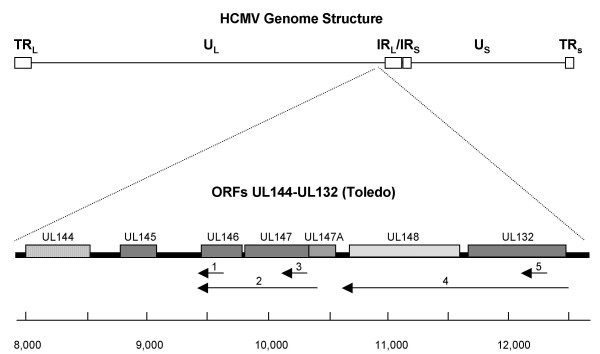
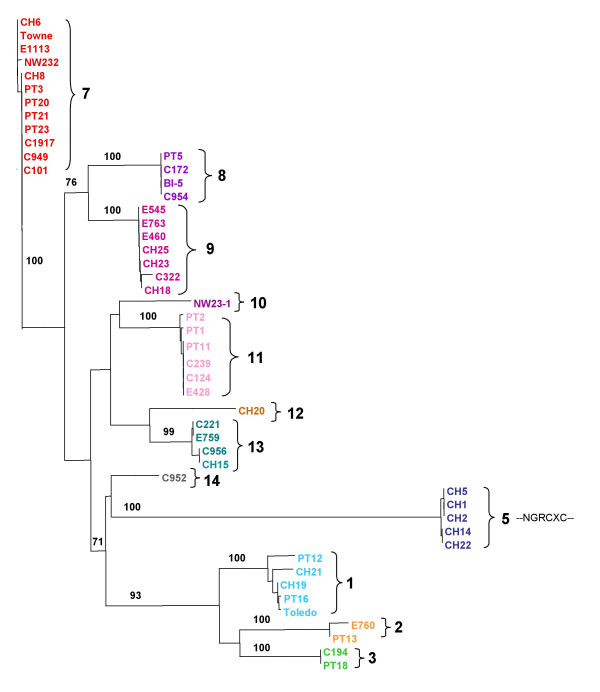
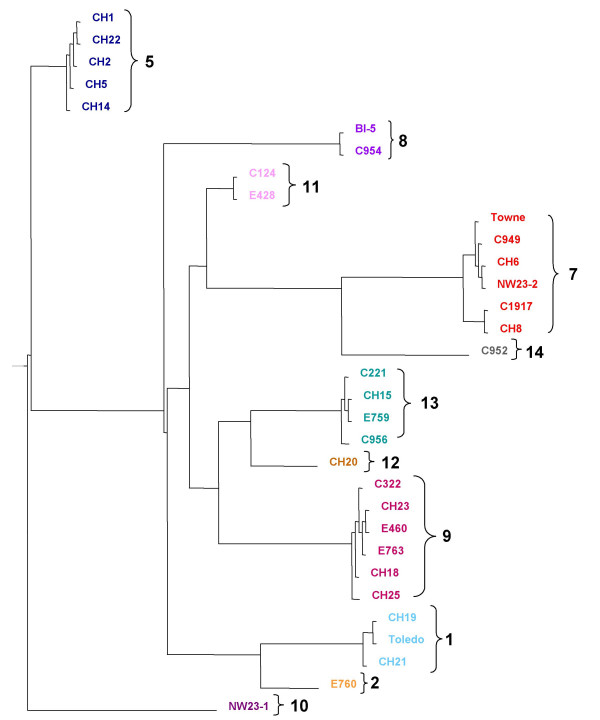
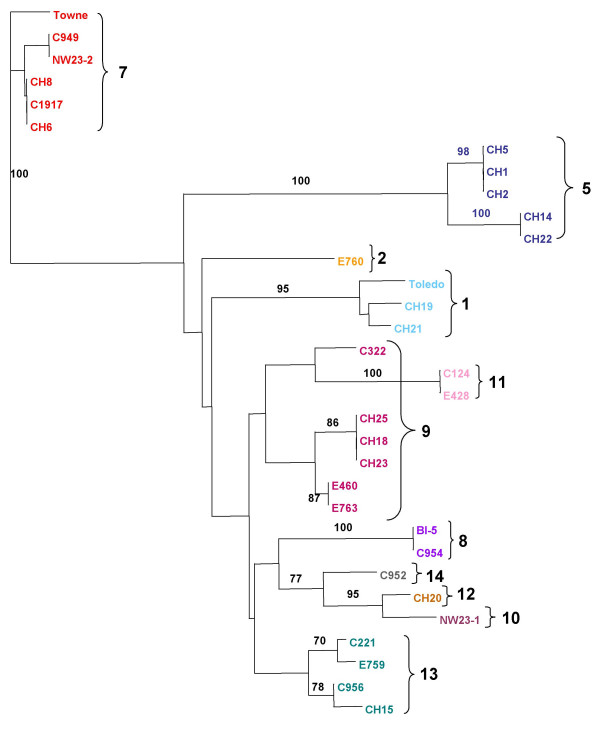
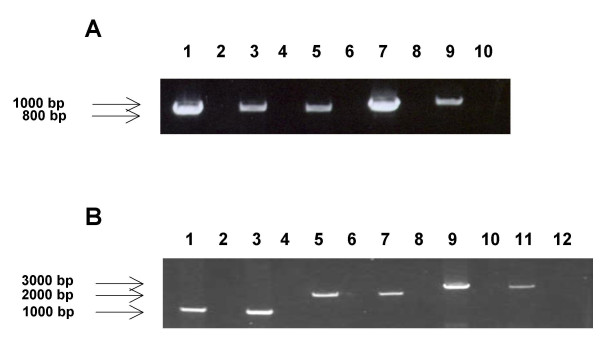
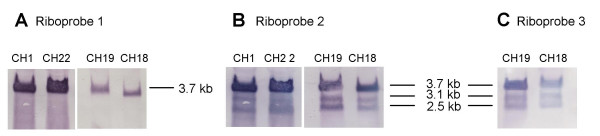
Results: The region of the HCMV genome from UL146 through UL147A was analyzed in clinical strains for sequence variability, genotypic stability, and transcriptional expression. The UL146 sequences in clinical strains from two geographically distant sites were assigned to 12 sequence groups that differ by over 60% at the amino acid level. The same groups were generated by sequences from the UL146-UL147 intergenic region and the UL147 ORF. In contrast to the high level of sequence variability among unrelated clinical strains, the sequences of UL146 through UL147A from isolates of the same strain were highly stable after repeated passage both in vitro and in vivo. Riboprobes homologous to these ORFs detected multiple overlapping transcripts differing in temporal expression. UL146 sequences are present only on the largest transcript, which also contains all of the downstream ORFs including UL148 and UL132. The sizes and hybridization patterns of the transcripts are consistent with a common 3'-terminus downstream of the UL132 ORF. Early-late expression of the transcripts associated with UL146 and UL147 is compatible with the potential role of CXC chemokines in pathogenesis associated with viral replication.
Conclusion: Clinical isolates from two different geographic sites cluster in the same groups based on the hypervariability of the UL146, UL147, or the intergenic sequences, which provides strong evidence for linkage and no evidence for interstrain recombination within this region. The sequence of individual strains was absolutely stable in vitro and in vivo, which indicates that sequence drift is not a mechanism for the observed sequence hypervariability. There is also no evidence of transcriptional splicing, although multiple overlapping transcripts extending into the adjacent UL148 and UL132 open reading frames were detected using gene-specific probes.
Figures

Similar articles
-
Sequence variability of human cytomegalovirus UL146 and UL147 genes in low-passage clinical isolates.Intervirology. 2006;49(4):215-23. doi: 10.1159/000091468. Epub 2006 Feb 16. Intervirology. 2006. PMID: 16491016
-
Characterization of human cytomegalovirus UL146 transcripts.Virus Res. 2012 Jan;163(1):223-8. doi: 10.1016/j.virusres.2011.09.034. Epub 2011 Oct 15. Virus Res. 2012. PMID: 22020362
-
UL146 variability among clinical isolates of human cytomegalovirus from Japan.Biol Res. 2010;43(4):475-80. Epub 2011 Feb 1. Biol Res. 2010. PMID: 21526275
-
[Cytomegalovirus].Nihon Rinsho. 2003 Mar;61 Suppl 3:587-93. Nihon Rinsho. 2003. PMID: 12718033 Review. Japanese. No abstract available.
-
Genetic polymorphisms among human cytomegalovirus (HCMV) wild-type strains.Rev Med Virol. 2004 Nov-Dec;14(6):383-410. doi: 10.1002/rmv.438. Rev Med Virol. 2004. PMID: 15386592 Review.
Cited by
-
Differential decay kinetics of human cytomegalovirus glycoprotein B genotypes following antiviral chemotherapy.J Clin Virol. 2012 May;54(1):56-60. doi: 10.1016/j.jcv.2012.01.015. Epub 2012 Mar 10. J Clin Virol. 2012. PMID: 22410132 Free PMC article.
-
Cytomegalovirus Genetic Diversity and Evolution: Insights into Genotypes and Their Role in Viral Pathogenesis.Pathogens. 2025 Jan 9;14(1):50. doi: 10.3390/pathogens14010050. Pathogens. 2025. PMID: 39861011 Free PMC article. Review.
-
The human cytomegalovirus protein UL147A downregulates the most prevalent MICA allele: MICA*008, to evade NK cell-mediated killing.PLoS Pathog. 2021 May 3;17(5):e1008807. doi: 10.1371/journal.ppat.1008807. eCollection 2021 May. PLoS Pathog. 2021. PMID: 33939764 Free PMC article.
-
Overlapping transcription structure of human cytomegalovirus UL140 and UL141 genes.J Biosci. 2013 Mar;38(1):35-44. doi: 10.1007/s12038-012-9293-4. J Biosci. 2013. PMID: 23385811
-
Ganciclovir-resistant cytomegalovirus infections among lung transplant recipients are associated with poor outcomes despite treatment with foscarnet-containing regimens.Antimicrob Agents Chemother. 2014;58(1):128-35. doi: 10.1128/AAC.00561-13. Epub 2013 Oct 21. Antimicrob Agents Chemother. 2014. PMID: 24145525 Free PMC article.
References
-
- Chee MS, Bankier AT, Beck S, Bohni R, Brown CM, Cerny R, Horsnell T, Hutchison CA, Kouzarides T, Martignetti JA, et al. Analysis of the protein-coding content of the sequence of human cytomegalovirus strain AD169. Curr Top Microbiol Immunol. 1990;154:125–169. - PubMed
-
- Dolan A, Cunningham C, Hector RD, Hassan-Walker AF, Lee L, Addison C, Dargan DJ, McGeoch DJ, Gatherer D, Emery VC, Griffiths PD, Sinzger C, McSharry BP, Wilkinson GW, Davison AJ. Genetic content of wild-type human cytomegalovirus. J Gen Virol. 2004;85:1301–1312. doi: 10.1099/vir.0.79888-0. - DOI - PubMed
-
- Chou SW, Dennison KM. Analysis of interstrain variation in cytomegalovirus glycoprotein B sequences encoding neutralization-related epitopes. J Infect Dis. 1991;163:1229–1234. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
