

Genome-wide identification of human functional DNA using a neutral indel model
- PMID: 16410828
- PMCID: PMC1326222
- DOI: 10.1371/journal.pcbi.0020005
Genome-wide identification of human functional DNA using a neutral indel model
Abstract
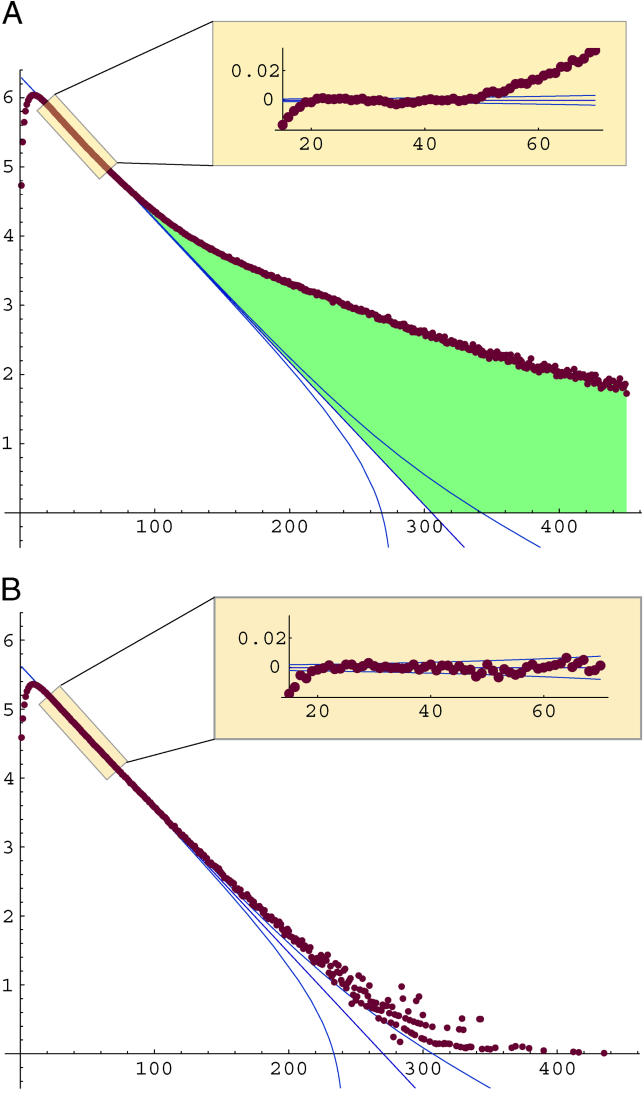
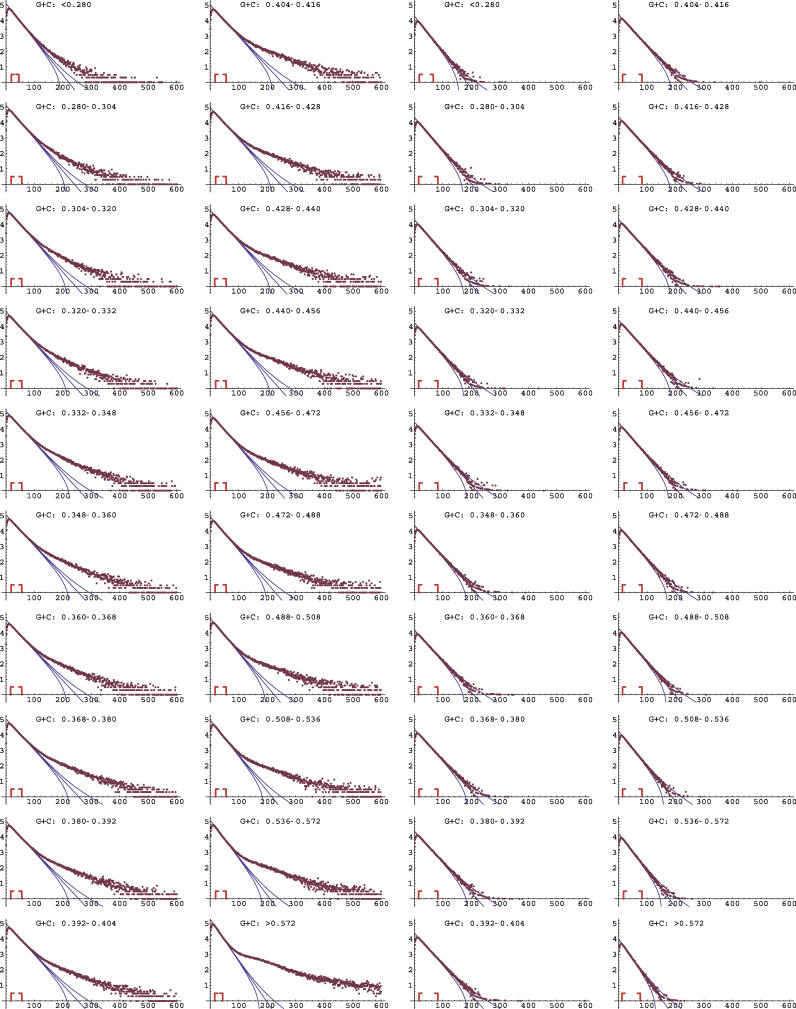
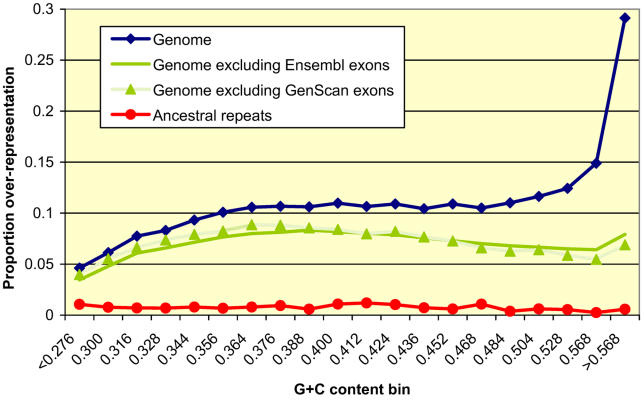
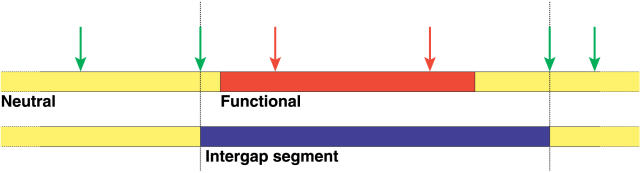
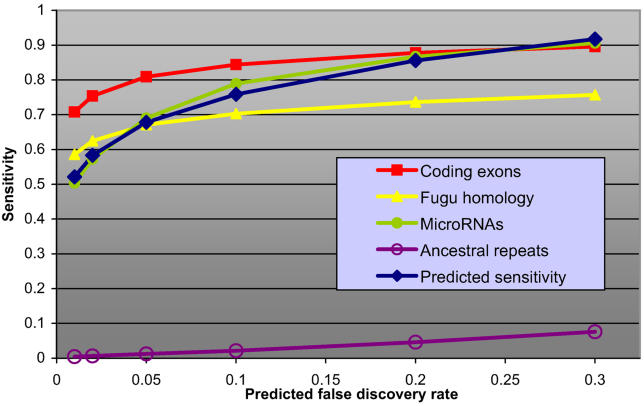
It has become clear that a large proportion of functional DNA in the human genome does not code for protein. Identification of this non-coding functional sequence using comparative approaches is proving difficult and has previously been thought to require deep sequencing of multiple vertebrates. Here we introduce a new model and comparative method that, instead of nucleotide substitutions, uses the evolutionary imprint of insertions and deletions (indels) to infer the past consequences of selection. The model predicts the distribution of indels under neutrality, and shows an excellent fit to human-mouse ancestral repeat data. Across the genome, many unusually long ungapped regions are detected that are unaccounted for by the neutral model, and which we predict to be highly enriched in functional DNA that has been subject to purifying selection with respect to indels. We use the model to determine the proportion under indel-purifying selection to be between 2.56% and 3.25% of human euchromatin. Since annotated protein-coding genes comprise only 1.2% of euchromatin, these results lend further weight to the proposition that more than half the functional complement of the human genome is non-protein-coding. The method is surprisingly powerful at identifying selected sequence using only two or three mammalian genomes. Applying the method to the human, mouse, and dog genomes, we identify 90 Mb of human sequence under indel-purifying selection, at a predicted 10% false-discovery rate and 75% sensitivity. As expected, most of the identified sequence represents unannotated material, while the recovered proportions of known protein-coding and microRNA genes closely match the predicted sensitivity of the method. The method's high sensitivity to functional sequence such as microRNAs suggest that as yet unannotated microRNA genes are enriched among the sequences identified. Furthermore, its independence of substitutions allowed us to identify sequence that has been subject to heterogeneous selection, that is, sequence subject to both positive selection with respect to substitutions and purifying selection with respect to indels. The ability to identify elements under heterogeneous selection enables, for the first time, the genome-wide investigation of positive selection on functional elements other than protein-coding genes.
Conflict of interest statement
Figures

References
-
- Mouse Genome Sequencing Consortium. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. - PubMed
-
- Chiaromonte F, Weber RJ, Roskin KM, Diekhans M, Kent WJ, et al. The share of human genomic DNA under selection estimated from human–mouse genomic alignments. Cold Spring Harb Symp Quant Biol. 2004;68:245–254. - PubMed
-
- International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931–945. - PubMed
-
- Bejerano G, Pheasant M, Makunin I, Stephen S, Kent WJ, et al. Ultraconserved Elements in the Human Genome. Science. 2004;304:1321–1325. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
