

Coronavirus infection of the central nervous system: host-virus stand-off
- PMID: 16415928
- PMCID: PMC7096820
- DOI: 10.1038/nrmicro1343
Coronavirus infection of the central nervous system: host-virus stand-off
Abstract
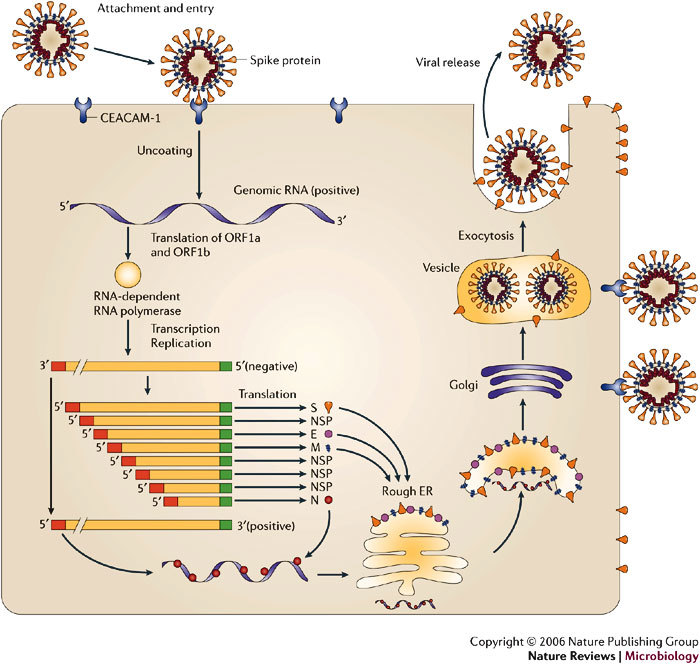
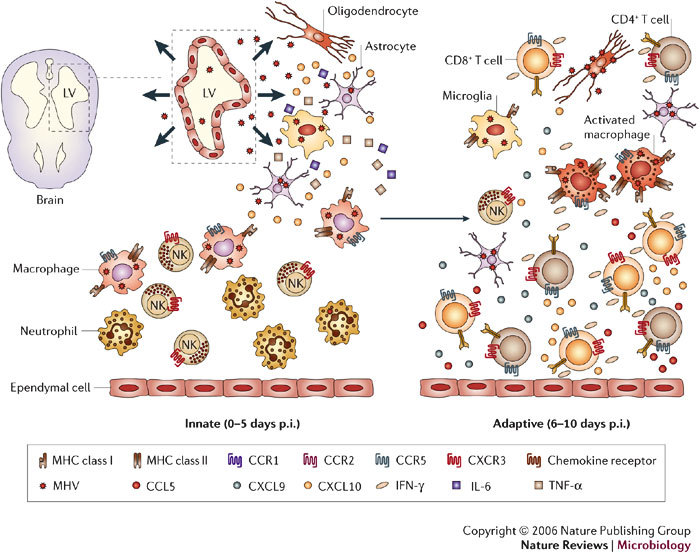
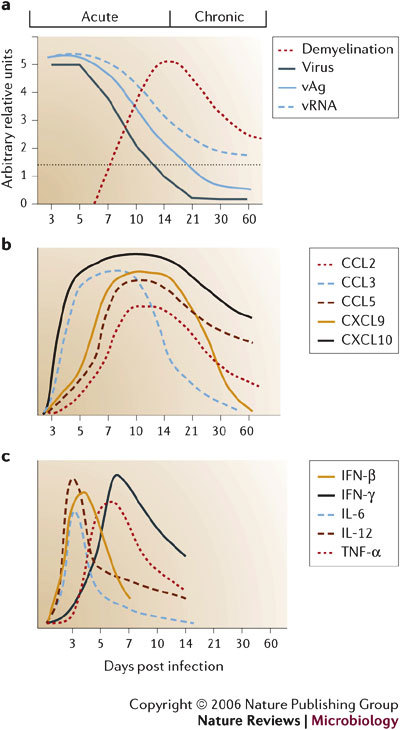
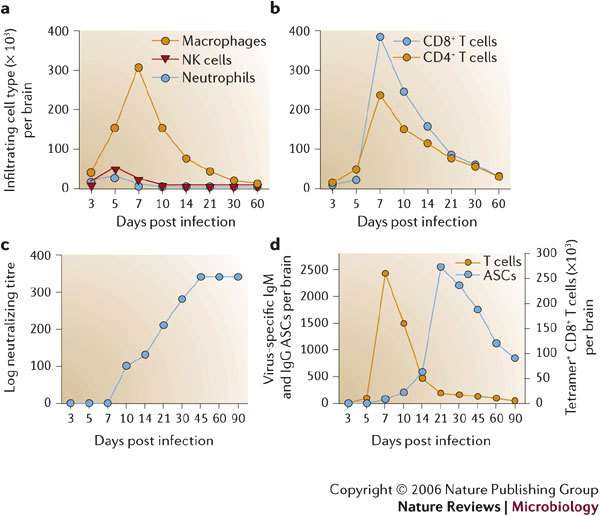
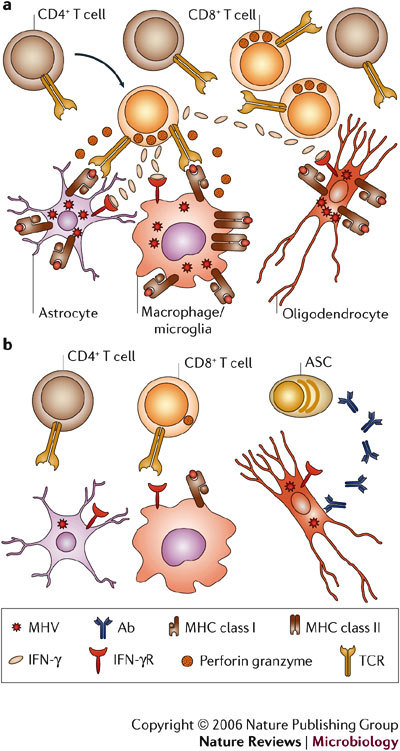
Several viruses infect the mammalian central nervous system (CNS), some with devastating consequences, others resulting in chronic or persistent infections associated with little or no overt pathology. Coronavirus infection of the murine CNS illustrates the contributions of both the innate immune response and specific host effector mechanisms that control virus replication in distinct CNS cell types. Despite T-cell-mediated control of acute virus infection, host regulatory mechanisms, probably designed to protect CNS integrity, contribute to the failure to eliminate virus. Distinct from cytolytic effector mechanisms expressed during acute infection, non-lytic humoral immunity prevails in suppressing infectious virus during persistence.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
-
- Fabry Z, Raine CS, Hart MN. Nervous tissue as an immune compartment: the dialect of the immune response in the CNS. Immunol. Today. 1994;15:218–224. - PubMed
-
- Hickey WF. Basic principles of immunological surveillance of the normal central nervous system. Glia. 2001;36:118–124. - PubMed
-
- Aloisi F, Ria F, Adorini L. Regulation of T-cell responses by CNS antigen-presenting cells: different roles for microglia and astrocytes. Immunol. Today. 2000;21:141–147. - PubMed
-
- Johnson MD, Gold LI, Moses HL. Evidence for transforming growth factor-β expression in human leptomeningeal cells and transforming growth factor-β-like activity in human cerebrospinal fluid. Lab. Invest. 1992;67:360–368. - PubMed
-
- Hoek RM, et al. Down-regulation of the macrophage lineage through interaction with OX2 (CD200) Science. 2000;290:1768–1771. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
