

doi: 10.1186/gb-2005-6-13-r114.
Epub 2005 Dec 19.
Discovery of biological networks from diverse functional genomic data
Affiliations
- PMID: 16420673
- PMCID: PMC1414113
- DOI: 10.1186/gb-2005-6-13-r114
Item in Clipboard
Discovery of biological networks from diverse functional genomic data
Genome Biol.
2005.
Abstract
We have developed a general probabilistic system for query-based discovery of pathway-specific networks through integration of diverse genome-wide data. This framework was validated by accurately recovering known networks for 31 biological processes in Saccharomyces cerevisiae and experimentally verifying predictions for the process of chromosomal segregation. Our system, bioPIXIE, a public, comprehensive system for integration, analysis, and visualization of biological network predictions for S. cerevisiae, is freely accessible over the worldwide web.
Figures
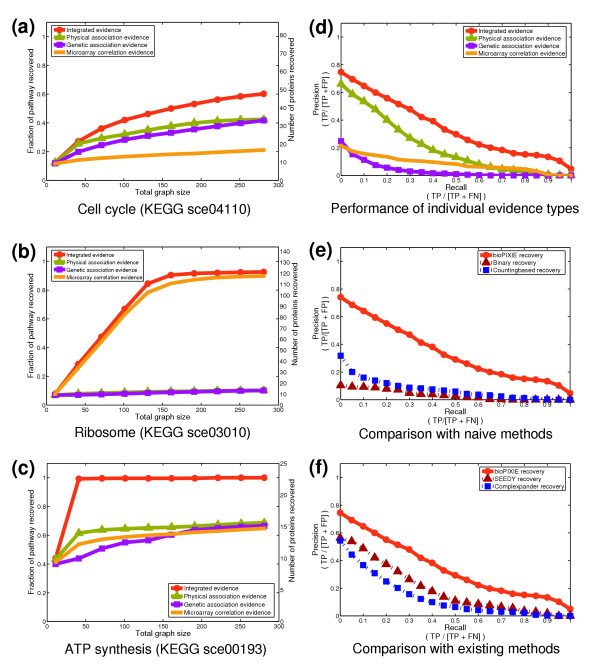
bioPIXIE network recovery evaluation. (a-c) Typical network recovery performance for three KEGG pathways. For all pathways, ten proteins from the pathway were randomly picked as a query set. The results of 100 independent query set samplings are shown. The fraction of the total known process components recovered is plotted versus the size of the graph grown from the query set. (d-f) An average over 31 KEGG pathways, GO biological processes, and MIPS complexes. Performance is measured and reported as the trade-off between precision (the proportion of correct pathway components returned to the total size of the returned network) and recall (the proportion of correct pathway components returned to the number of total non-query pathway proteins). Precision and recall are derived from true positives (TP), false positives (FP), and false negatives (FN) as noted in the axis labels. (d) The improvement gained by using our network prediction algorithm on a Bayesian integration of genomic evidence compared to separate evidence types. bioPIXIE shows considerable improvement in both the number of known member proteins recovered and the precision of predicted members for the integrated evidence over any individual evidence type. (e) The improved network recovery offered by the bioPIXIE algorithm versus more naïve approaches to integration and graph search. Specifically, we plot the performance of bioPIXIE on integrated data against a naïve binary approach for which information from all evidence types is used but only as a binary 'yes' or 'no' relationship, and a more sophisticated approach where overlapping evidence receives higher weights and connected proteins are recovered in order of confidence. (f) Comparison of the performance of bioPIXIE to two existing methods for query-based protein complex recovery [13,14].
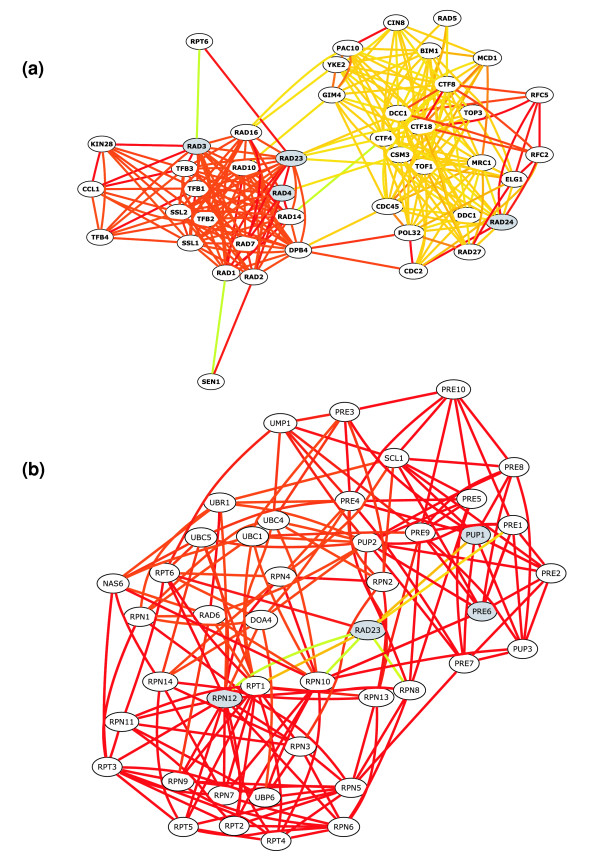
bioPIXIE query-driven context illustration. Nodes represent proteins, and edges represent functional links between them. Edge color indicates the confidence of the links ordered by color from red (highest confidence), orange, yellow, to green (lowest confidence). Query proteins are indicated by gray nodes. Rad23 is known to form a complex with Rad4 (NEF2) and participate in nucleotide excision repair and has also been implicated in inhibiting the degradation of specific substrates in response to DNA damage. (a) Rad23 was entered with Rad4, Rad3, and Rad24 and the resulting network is enriched (22 of 44, P value < 10-22) for DNA repair proteins (GO:0006281). (b) Rad23 was entered with proteasome components Pup1, Pre6, Rpn12 and the recovered network is enriched (36 of 44, P value < 10-55) for ubiquitin-dependent catabolism proteins (GO:0006511) and only contains 2 DNA repair proteins (Rad6 and Rad23). Rad23 has high-confidence relationships with several proteins in both processes, but the network recovery algorithm is dependent on the context of the query, which results in two different views of Rad23 and its neighbors.

Experimental validation of bioPIXIE prediction for the biological role of YPL017C. bioPIXIE was used to predict previously uncharacterized genes likely to participate in processes related to chromosomal segregation (data for YPL017C shown). Yeast cells were fixed, stained, and photographed using differential interference contrast imaging and 4'-6-diamidino-2-phenylindole (DAPI) staining. When compared with wild-type cells, populations of cells lacking YPL017C have a higher proportion of large-budded cells with a single nucleus at the bud neck (75% compared to 22% in wild type, Fisher exact test P value of 5 × 10-9). Large budding cells are indicated by arrows. This morphology and failure of nuclear separation are analogous to that of ctf4Δ mutants [19], supporting the hypothesis that YPL017C, like CTF4, is involved in chromosome segregation. See Figure S8 in [15] for experimental verification of YPL077C and YPL144W.
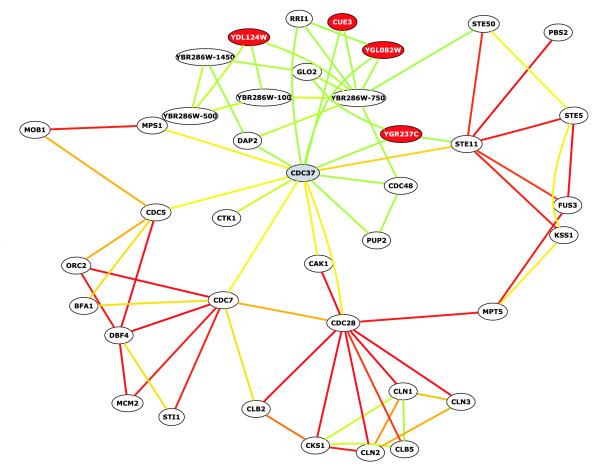
bioPIXIE output for Cdc37. Nodes represent genes, and edges represent functional links between them. Edge color indicates the confidence of the links ordered by color, from red (highest confidence), orange, yellow, to green (lowest confidence). In this example, CDC37 was entered as input (gray node); other genes displayed (white nodes) were identified by the bioPIXIE prediction algorithm. Red nodes indicate that the gene is uncharacterized. These results and networks for other proteins can be viewed at [54].
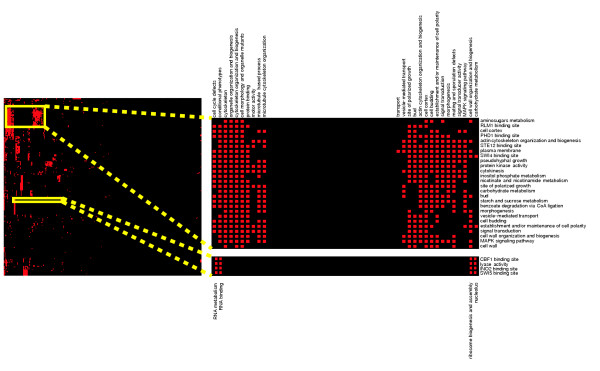
A map of cross-talk between 363 biological groups in S. cerevisiae. The combination of our Bayesian data integration system and our network discovery algorithm allows us to find biologically significant cross-talk among known biological groups. The interaction matrix was generated based on 363 KEGG pathways, GO categories, and co-regulated transcription factor targets. Rows of this matrix correspond to the query group and columns correspond to potential cross-talk partner processes; red boxes signify statistically significant links. The cross-talk matrix has been clustered [58] to reveal tightly connected groups of interacting processes (clusters in this matrix correspond to sets of groups who interact with same partners). Highlighted clusters are discussed in the text. See supplemental Figure S10 in [15] for a complete, labeled map.
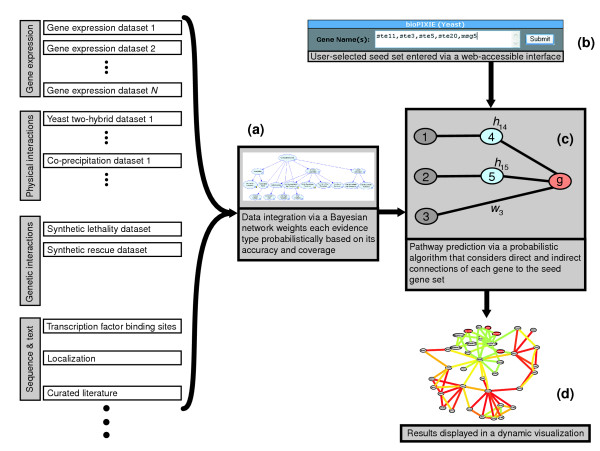
Overview of the bioPIXIE system. Diverse data sets are integrated with a Bayesian network, which weighs each evidence type probabilistically based on its accuracy (a). This Bayesian integration produces a graph with confidence-weighted relationships between each gene pair (characterized in supplemental Figure S1 in [15]). Based on this integrated network graph and a user-defined query set of proteins of interest (b), the network prediction algorithm identifies novel network components by finding proteins with the maximum expected number of direct and indirect relationships with the query set (c). The resulting network is then displayed to the user using a spring model layout, such that the geometric proximity of genes reflects how related they are to each other, and the edge color reflects the confidence of pair-wise connections (d). Details of each component are presented in Materials and methods.
References
-
- Jaimovich A, Elidan G, Margalit H, Friedman N. Towards an integrated protein-protein interaction network. In: Miyano S, Mesirov J, Kasif S, Istrail S, Pevzner P, Waterman M, editor. Research in Computational Molecular Biology: 9th Annual International Conference, RECOMB, Proceedings: May 14-18 2005, Cambridge, MA. Springer Verlag-GmbH; 2005. pp. 14–30.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
