

A rivet model for channel formation by aerolysin-like pore-forming toxins
- PMID: 16424900
- PMCID: PMC1383540
- DOI: 10.1038/sj.emboj.7600959
A rivet model for channel formation by aerolysin-like pore-forming toxins
Abstract
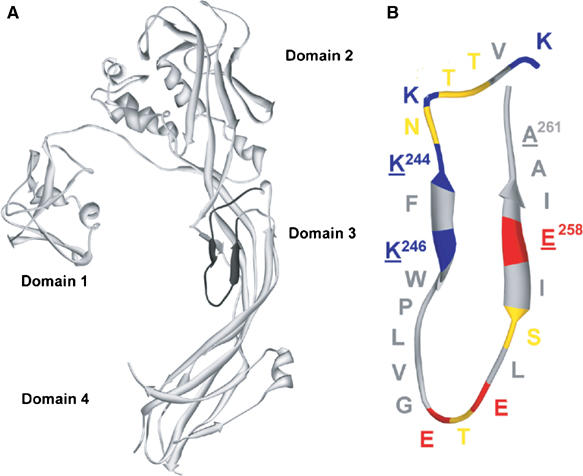
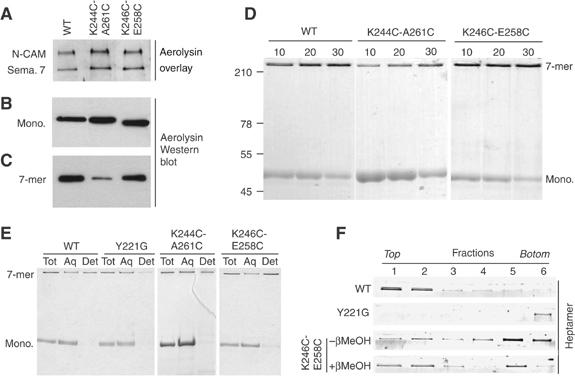
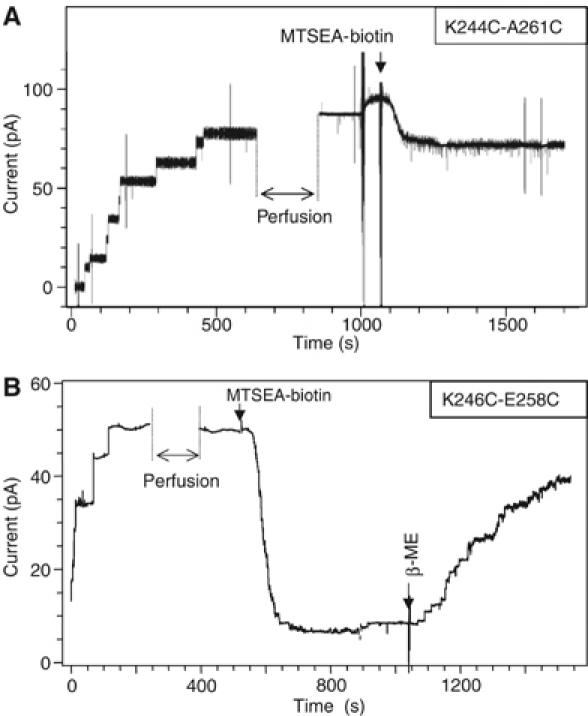
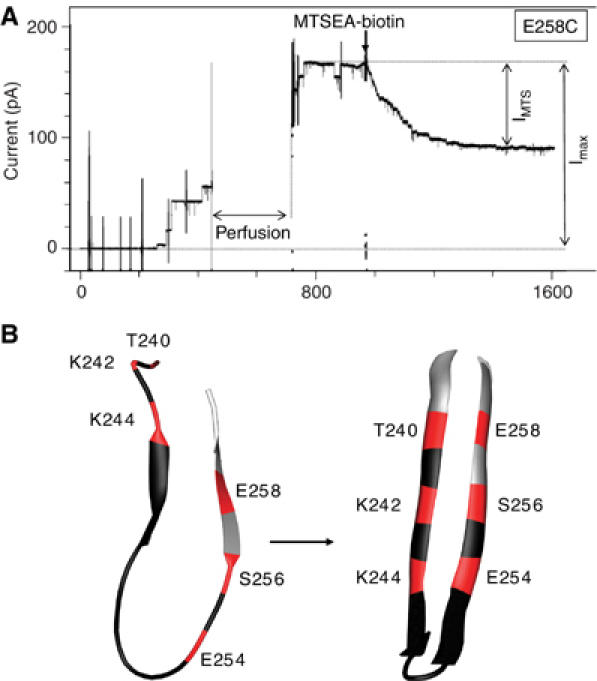
The bacterial toxin aerolysin kills cells by forming heptameric channels, of unknown structure, in the plasma membrane. Using disulfide trapping and cysteine scanning mutagenesis coupled to thiol-specific labeling on lipid bilayers, we identify a loop that lines the channel. This loop has an alternating pattern of charged and uncharged residues, suggesting that the transmembrane region has a beta-barrel configuration, as observed for Staphylococcal alpha-toxin. Surprisingly, we found that the turn of the beta-hairpin is composed of a stretch of five hydrophobic residues. We show that this hydrophobic turn drives membrane insertion of the developing channel and propose that, once the lipid bilayer has been crossed, it folds back parallel to the plane of the membrane in a rivet-like fashion. This rivet-like conformation was modeled and sequence alignments suggest that such channel riveting may operate for many other pore-forming toxins.
Figures


Similar articles
-
Aerolysin--a paradigm for membrane insertion of beta-sheet protein toxins?J Struct Biol. 1998;121(2):92-100. doi: 10.1006/jsbi.1997.3947. J Struct Biol. 1998. PMID: 9615432 Review.
-
Molecular architecture of a toxin pore: a 15-residue sequence lines the transmembrane channel of staphylococcal alpha-toxin.EMBO J. 1996 Apr 15;15(8):1857-64. EMBO J. 1996. PMID: 8617232 Free PMC article.
-
Clostridium perfringens epsilon-toxin shows structural similarity to the pore-forming toxin aerolysin.Nat Struct Mol Biol. 2004 Aug;11(8):797-8. doi: 10.1038/nsmb804. Epub 2004 Jul 18. Nat Struct Mol Biol. 2004. PMID: 15258571
-
Movement of a loop in domain 3 of aerolysin is required for channel formation.Biochemistry. 1998 Jan 13;37(2):741-6. doi: 10.1021/bi9721039. Biochemistry. 1998. PMID: 9425098
-
Surface dynamics of aerolysin on the plasma membrane of living cells.Int J Med Microbiol. 2000 Oct;290(4-5):363-7. doi: 10.1016/S1438-4221(00)80042-9. Int J Med Microbiol. 2000. PMID: 11111912 Review.
Cited by
-
Biomphalysin, a new β pore-forming toxin involved in Biomphalaria glabrata immune defense against Schistosoma mansoni.PLoS Pathog. 2013 Mar;9(3):e1003216. doi: 10.1371/journal.ppat.1003216. Epub 2013 Mar 21. PLoS Pathog. 2013. PMID: 23555242 Free PMC article.
-
Morin Protects Channel Catfish From Aeromonas hydrophila Infection by Blocking Aerolysin Activity.Front Microbiol. 2018 Nov 21;9:2828. doi: 10.3389/fmicb.2018.02828. eCollection 2018. Front Microbiol. 2018. PMID: 30519232 Free PMC article.
-
Patch repair protects cells from the small pore-forming toxin aerolysin.J Cell Sci. 2023 Apr 15;136(8):jcs261018. doi: 10.1242/jcs.261018. Epub 2023 Apr 20. J Cell Sci. 2023. PMID: 36951121 Free PMC article.
-
The pore structure of Clostridium perfringens epsilon toxin.Nat Commun. 2019 Jun 14;10(1):2641. doi: 10.1038/s41467-019-10645-8. Nat Commun. 2019. PMID: 31201325 Free PMC article.
-
Preliminary crystallographic analysis of two oligomerization-deficient mutants of the aerolysin toxin, H132D and H132N, in their proteolyzed forms.Acta Crystallogr Sect F Struct Biol Cryst Commun. 2010 Dec 1;66(Pt 12):1626-30. doi: 10.1107/S1744309110041035. Epub 2010 Nov 26. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2010. PMID: 21139211 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
