

Mouse models in non-alcoholic fatty liver disease and steatohepatitis research
- PMID: 16436109
- PMCID: PMC2517349
- DOI: 10.1111/j.0959-9673.2006.00465.x
Mouse models in non-alcoholic fatty liver disease and steatohepatitis research
Abstract
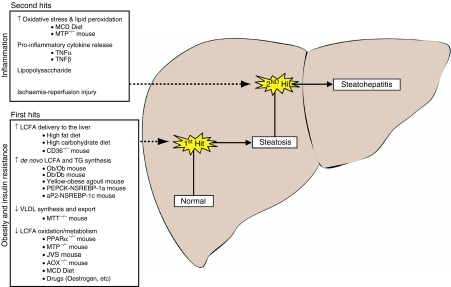
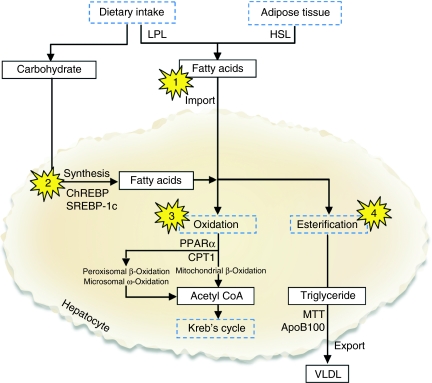
Non-alcoholic fatty liver disease (NAFLD) represents a histological spectrum of liver disease associated with obesity, diabetes and insulin resistance that extends from isolated steatosis to steatohepatitis and cirrhosis. As well as being a potential cause of progressive liver disease in its own right, steatosis has been shown to be an important cofactor in the pathogenesis of many other liver diseases. Animal models of NAFLD may be divided into two broad categories: those caused by genetic mutation and those with an acquired phenotype produced by dietary or pharmacological manipulation. The literature contains numerous different mouse models that exhibit histological evidence of hepatic steatosis or, more variably, steatohepatitis; however, few replicate the entire human phenotype. The genetic leptin-deficient (ob/ob) or leptin-resistant (db/db) mouse and the dietary methionine/choline-deficient model are used in the majority of published research. More recently, targeted gene disruption and the use of supra-nutritional diets to induce NAFLD have gained greater prominence as researchers have attempted to bridge the phenotype gap between the available models and the human disease. Using the physiological processes that underlie the pathogenesis and progression of NAFLD as a framework, we review the literature describing currently available mouse models of NAFLD, highlight the strengths and weaknesses of established models and describe the key findings that have furthered the understanding of disease pathogenesis.
Figures
References
-
- Adinolfi LE, Gambardella M, Andreana A, Tripodi MF, Utili R, Ruggiero G. Steatosis accelerates the progression of liver damage of chronic hepatitis C patients and correlates with specific HCV genotype and visceral obesity. Hepatology. 2001;33:1358–1364. - PubMed
-
- Ahima RS, Saper CB, Flier JS, Elmquist JK. Leptin regulation of neuroendocrine systems. Front Neuroendocrinol. 2000;21:263–307. - PubMed
-
- Almind K, Doria A, Kahn CR. Putting the genes for type II diabetes on the map. Nat. Med. 2001;7:277–279. - PubMed
-
- Angulo P, Lindor KD. Treatment of non-alcoholic steatohepatitis. Best Pract. Res. Clin. Gastroenterol. 2002;16x:797–810. - PubMed
-
- Anstee QM, Wright M, Goldin R, et al. A Novel murine model of non-alcoholic steatohepatitis generated using a sensitised ENU mutagenesis screen. Hepatology. 2003;1(Supplement):469A.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
