

TRPM7 regulates cell adhesion by controlling the calcium-dependent protease calpain
- PMID: 16436382
- PMCID: PMC3225339
- DOI: 10.1074/jbc.M512885200
TRPM7 regulates cell adhesion by controlling the calcium-dependent protease calpain
Abstract
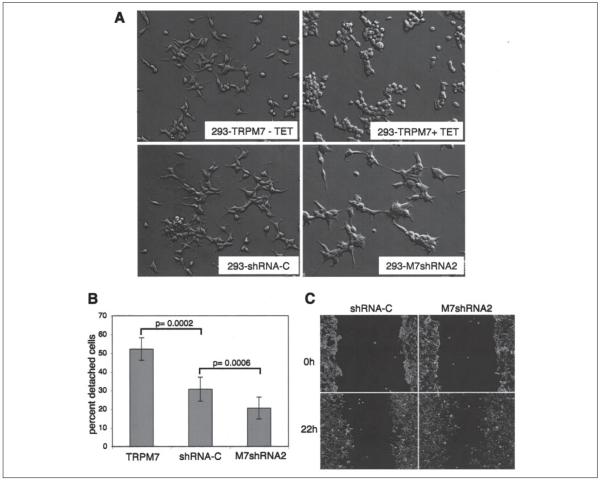
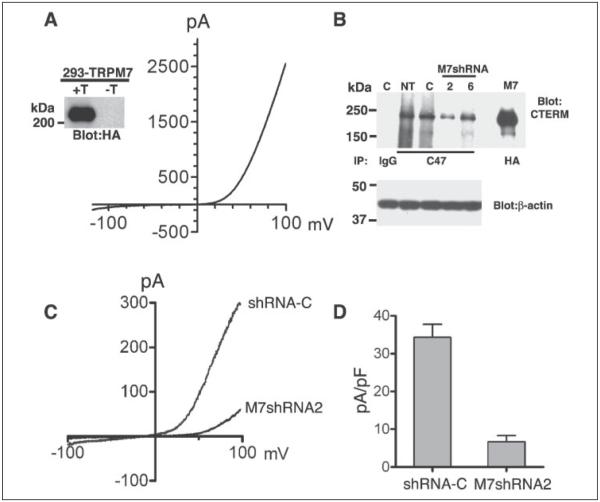
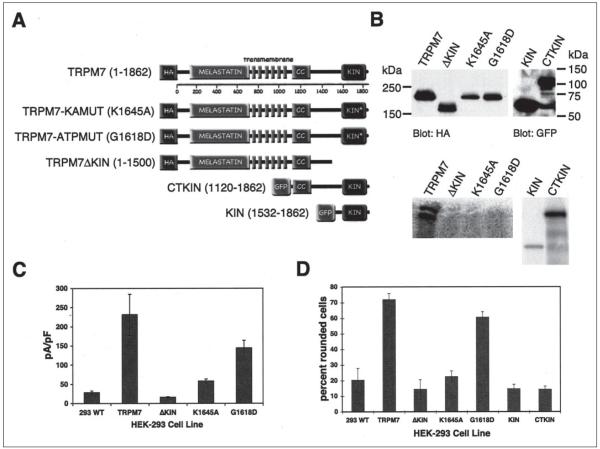
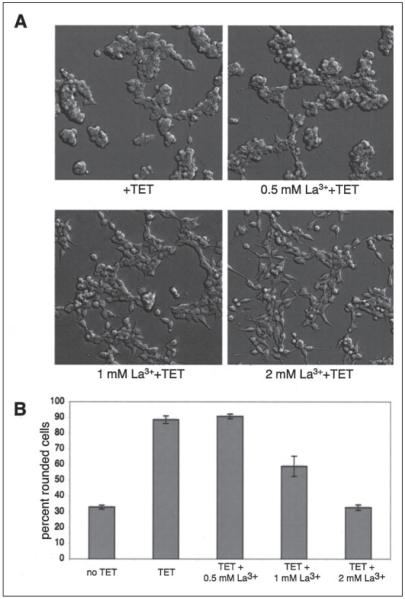
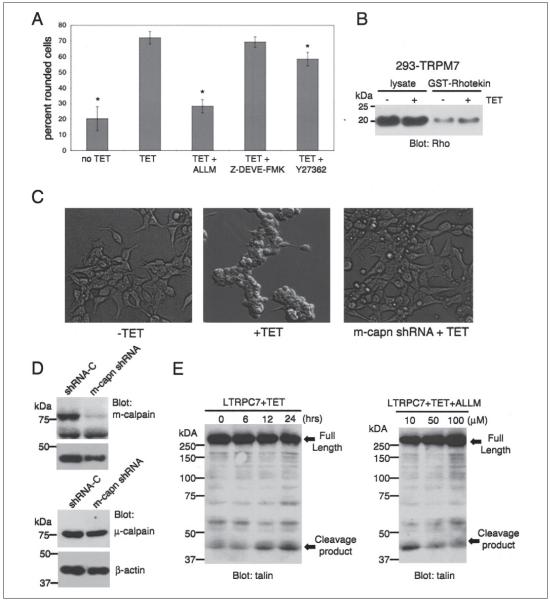
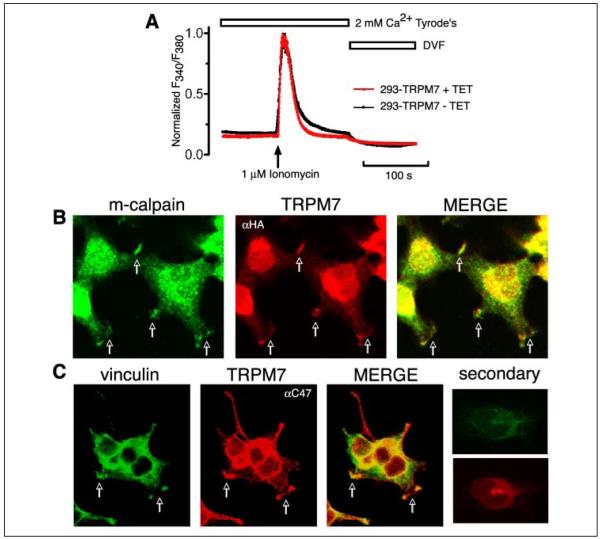
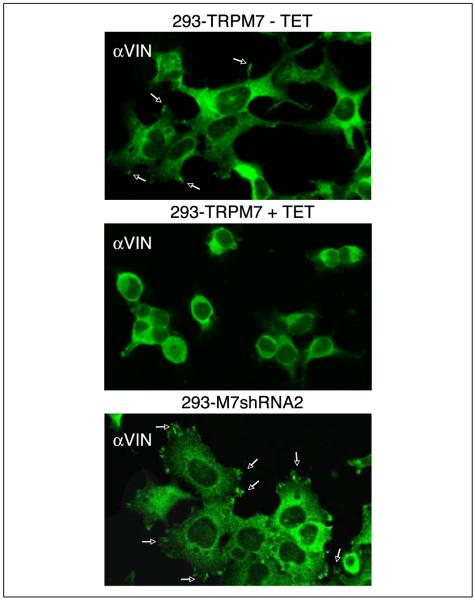
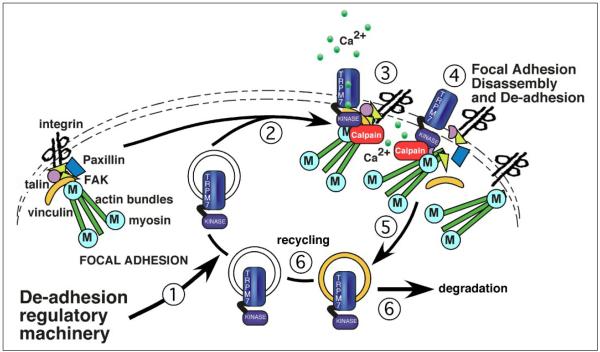
m-Calpain is a protease implicated in the control of cell adhesion through focal adhesion disassembly. The mechanism by which the enzyme is spatially and temporally controlled is not well understood, particularly because the dependence of calpain on calcium exceeds the submicromolar concentrations normally observed in cells. Here we show that the channel kinase TRPM7 localizes to peripheral adhesion complexes with m-calpain, where it regulates cell adhesion by controlling the activity of the protease. Our research revealed that overexpression of TRPM7 in cells caused cell rounding with a concomitant loss of cell adhesion that is dependent upon the channel of the protein but not its kinase activities. Knockdown of m-calpain blocked TRPM7-induced cell rounding and cell detachment. Silencing of TRPM7 by RNA interference, however, strengthened cell adhesion and increased the number of peripheral adhesion complexes in the cells. Together, our results suggest that the ion channel TRPM7 regulates cell adhesion through m-calpain by mediating the local influx of calcium into peripheral adhesion complexes.
Figures
References
-
- Runnels LW, Yue L, Clapham DE. Science. 2001;291:1043–1047. - PubMed
-
- Fleig A, Penner R. Novartis Found. Symp. 2004;258:248–266. - PubMed
-
- Nilius B, Voets T. Novartis Found Symp. 2004;258:140–159. 263–266. - PubMed
-
- Clapham DE. Nature. 2003;426:517–524. - PubMed
-
- Montell C, Birnbaumer L, Flockerzi V. Cell. 2002;108:595–598. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
