

Potencies of human immunodeficiency virus protease inhibitors in vitro against Plasmodium falciparum and in vivo against murine malaria
- PMID: 16436721
- PMCID: PMC1366900
- DOI: 10.1128/AAC.50.2.639-648.2006
Potencies of human immunodeficiency virus protease inhibitors in vitro against Plasmodium falciparum and in vivo against murine malaria
Abstract
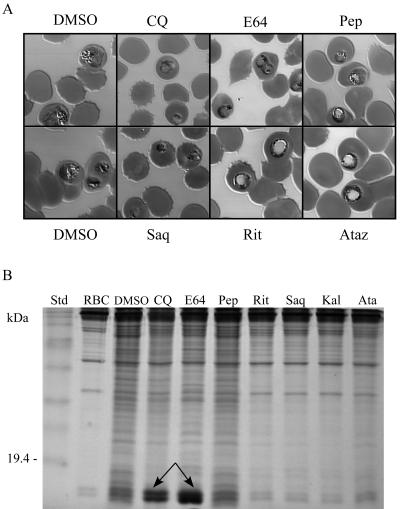
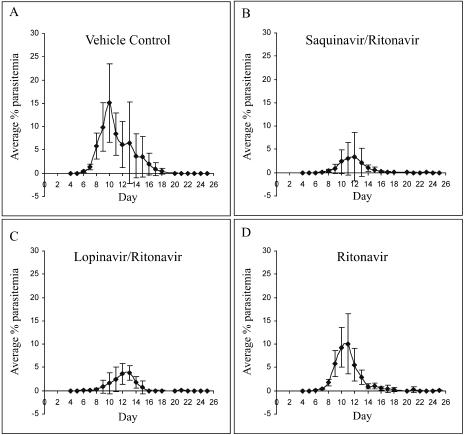
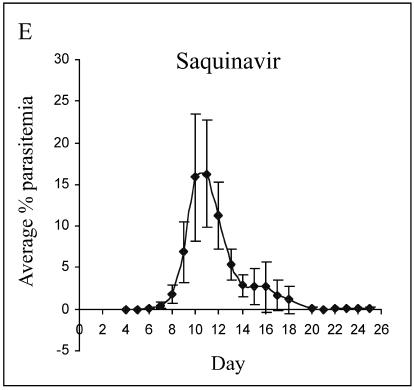
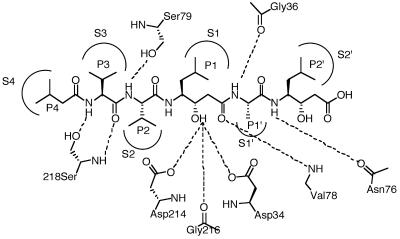
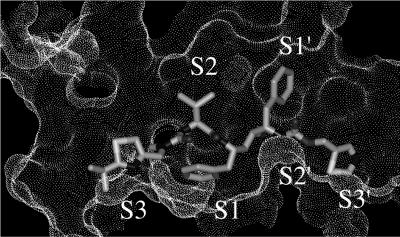
Parasite resistance to antimalarial drugs is a serious threat to human health, and novel agents that act on enzymes essential for parasite metabolism, such as proteases, are attractive targets for drug development. Recent studies have shown that clinically utilized human immunodeficiency virus (HIV) protease inhibitors can inhibit the in vitro growth of Plasmodium falciparum at or below concentrations found in human plasma after oral drug administration. The most potent in vitro antimalarial effects have been obtained for parasites treated with saquinavir, ritonavir, or lopinavir, findings confirmed in this study for a genetically distinct P. falciparum line (3D7). To investigate the potential in vivo activity of antiretroviral protease inhibitors (ARPIs) against malaria, we examined the effect of ARPI combinations in a murine model of malaria. In mice infected with Plasmodium chabaudi AS and treated orally with ritonavir-saquinavir or ritonavir-lopinavir, a delay in patency and a significant attenuation of parasitemia were observed. Using modeling and ligand docking studies we examined putative ligand binding sites of ARPIs in aspartyl proteases of P. falciparum (plasmepsins II and IV) and P. chabaudi (plasmepsin) and found that these in silico analyses support the antimalarial activity hypothesized to be mediated through inhibition of these enzymes. In addition, in vitro enzyme assays demonstrated that P. falciparum plasmepsins II and IV are both inhibited by the ARPIs saquinavir, ritonavir, and lopinavir. The combined results suggest that ARPIs have useful antimalarial activity that may be especially relevant in geographical regions where HIV and P. falciparum infections are both endemic.
Figures

References
-
- Abbenante, G., and D. P. Fairlie. 2005. Protease inhibitors in the clinic. Med. Chem. 1:71-104. - PubMed
-
- Andrews, K. T., A. Walduck, M. J. Kelso, D. P. Fairlie, A. Saul, and P. G. Parsons. 2000. Anti-malarial effect of histone deacetylation inhibitors and mammalian tumour cytodifferentiating agents. Int. J. Parasitol. 30:761-768. - PubMed
-
- Asojo, O. A., S. V. Gulnik, E. Afonina, B. Yu, J. A. Ellman, T. S. Haque, and A. M. Silva. 2003. Novel uncomplexed and complexed structures of plasmepsin II, an aspartic protease from Plasmodium falciparum. J. Mol. Biol. 327:173-181. - PubMed
-
- Bailey, E., R. Jambou, J. Savel, and G. Jaureguiberry. 1992. Plasmodium falciparum: differential sensitivity in vitro to E-64 (cysteine protease inhibitor) and pepstatin A (aspartyl protease inhibitor). J. Protozool. 39:593-599. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
