

High-density lipoprotein hydrolysis by endothelial lipase activates PPARalpha: a candidate mechanism for high-density lipoprotein-mediated repression of leukocyte adhesion
- PMID: 16439686
- PMCID: PMC4231787
- DOI: 10.1161/01.RES.0000205846.46812.be
High-density lipoprotein hydrolysis by endothelial lipase activates PPARalpha: a candidate mechanism for high-density lipoprotein-mediated repression of leukocyte adhesion
Abstract
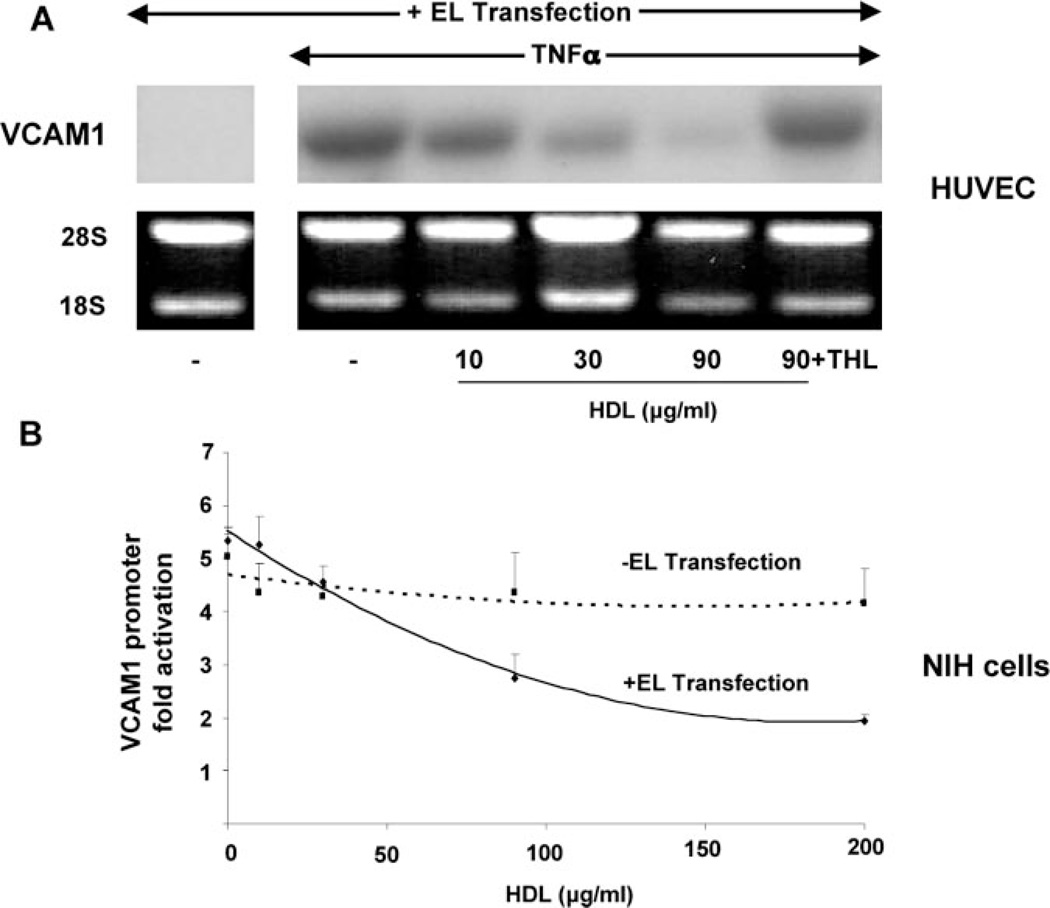
Although high-density lipoprotein (HDL) is known to inhibit endothelial adhesion molecule expression, the mechanism for this anti-inflammatory effect remains obscure. Surprisingly, we observed that HDL no longer decreased adhesion of U937 monocytoid cells to tumor necrosis factor (TNF)alpha-stimulated human endothelial cells (EC) in the presence of the general lipase inhibitor tetrahydrolipstatin. In considering endothelial mechanisms responsible for this effect, we found that endothelial lipase (EL) overexpression in both EC and non-EL-expressing NIH/3T3 mouse embryonic fibroblasts cells significantly decreased TNFalpha-induced VCAM1 expression and promoter activity in a manner dependent on HDL concentration and intact EL activity. Given recent evidence for lipolytic activation of peroxisome proliferator-activated receptors (PPARs)-nuclear receptors implicated in metabolism, atherosclerosis, and inflammation-we hypothesized HDL hydrolysis by EL is an endogenous endothelial mechanism for PPAR activation. In both EL-transfected NIH cells and bovine EC, HDL significantly increased PPAR ligand binding domain activation in the order PPAR-alpha> >-gamma>-delta. Moreover, HDL stimulation induced expression of the canonical PPARalpha-target gene acyl-CoA-oxidase (ACO) in a PPARalpha-dependent manner in ECs. Conditioned media from EL-adenovirus transfected cells but not control media exposed to HDL also activated PPARalpha. PPARalpha activation by EL was most potent with HDL as a substrate, with lesser effects on LDL and VLDL. Finally, HDL inhibited leukocyte adhesion to TNFalpha-stimulated ECs isolated from wild-type but not PPARalpha-deficient mice. This data establishes HDL hydrolysis by EL as a novel, distinct natural pathway for PPARalpha activation and identifies a potential mechanism for HDL-mediated repression of VCAM1 expression, with significant implications for both EL and PPARs in inflammation and vascular biology.
Figures







References
-
- Gordon T, Castelli WP, Hjortland MC, Kannel WB, Dawber TR. High density lipoprotein as a protective factor against coronary heart disease. The Framingham Study. Am J Med. 1977;62:707–714. - PubMed
-
- Castelli WP, Garrison RJ, Wilson PW, Abbott RD, Kalousdian S, Kannel WB. Incidence of coronary heart disease and lipoprotein cholesterol levels. The Framingham Study. JAMA. 1986;256:2835–2838. - PubMed
-
- Glass CK, Witztum JL. Atherosclerosis. the road ahead. Cel. 2001;104:503–516. - PubMed
-
- Cockerill GW, Rye KA, Gamble JR, Vadas MA, Barter PJ. High-density lipoproteins inhibit cytokine-induced expression of endothelial cell adhesion molecules. Arterioscler Thromb Vasc Biol. 1995;15:1987–1994. - PubMed
-
- Calabresi L, Franceschini G, Sirtori CR, De Palma A, Saresella M, Ferrante P, Taramelli D. Inhibition of VCAM-1 expression in endothelial cells by reconstituted high density lipoproteins. Biochem Biophys Res Commun. 1997;238:61–65. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
