

Computational analysis and prediction of the binding motif and protein interacting partners of the Abl SH3 domain
- PMID: 16446784
- PMCID: PMC1356089
- DOI: 10.1371/journal.pcbi.0020001
Computational analysis and prediction of the binding motif and protein interacting partners of the Abl SH3 domain
Abstract
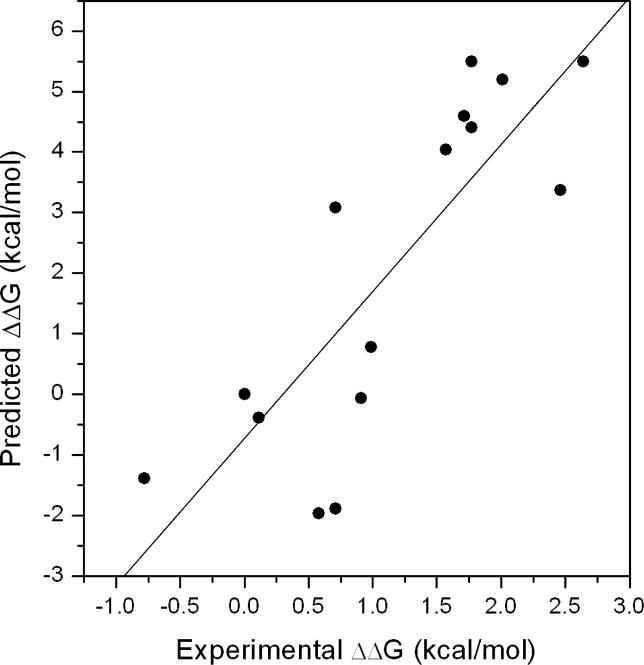
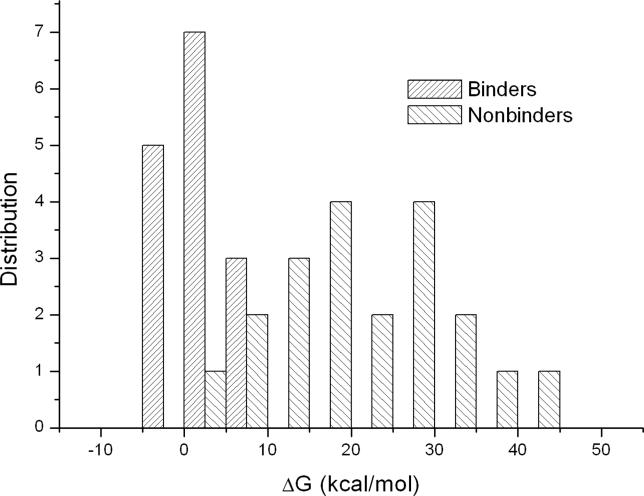
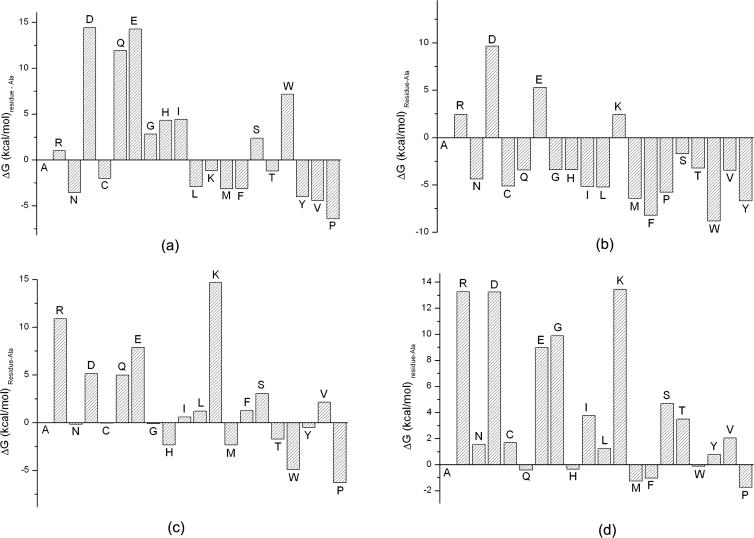
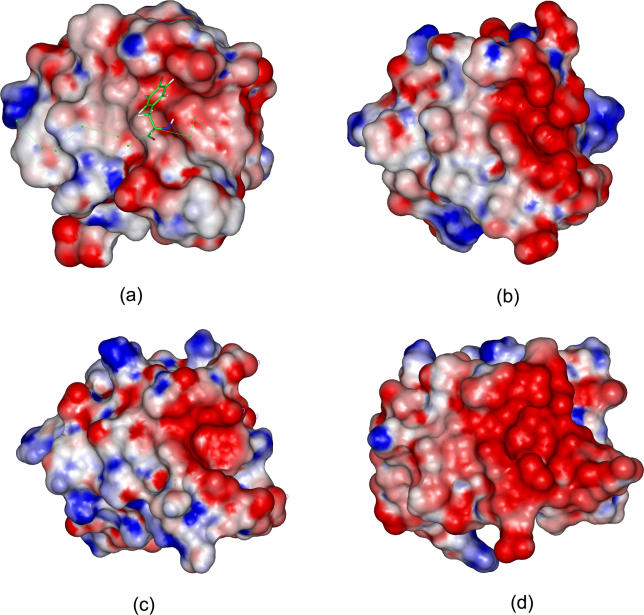
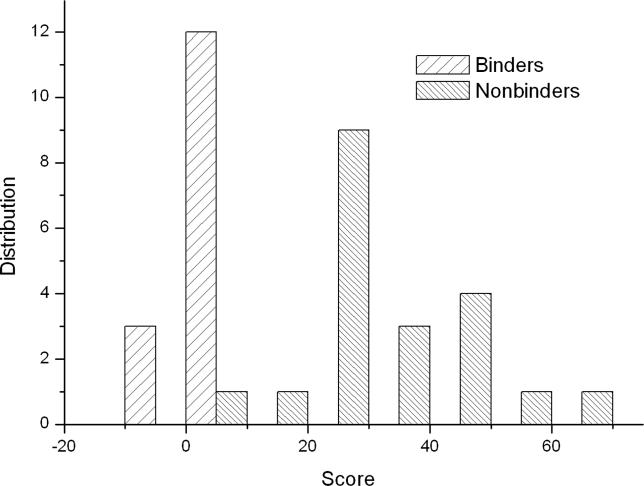
Protein-protein interactions, particularly weak and transient ones, are often mediated by peptide recognition domains, such as Src Homology 2 and 3 (SH2 and SH3) domains, which bind to specific sequence and structural motifs. It is important but challenging to determine the binding specificity of these domains accurately and to predict their physiological interacting partners. In this study, the interactions between 35 peptide ligands (15 binders and 20 non-binders) and the Abl SH3 domain were analyzed using molecular dynamics simulation and the Molecular Mechanics/Poisson-Boltzmann Solvent Area method. The calculated binding free energies correlated well with the rank order of the binding peptides and clearly distinguished binders from non-binders. Free energy component analysis revealed that the van der Waals interactions dictate the binding strength of peptides, whereas the binding specificity is determined by the electrostatic interaction and the polar contribution of desolvation. The binding motif of the Abl SH3 domain was then determined by a virtual mutagenesis method, which mutates the residue at each position of the template peptide relative to all other 19 amino acids and calculates the binding free energy difference between the template and the mutated peptides using the Molecular Mechanics/Poisson-Boltzmann Solvent Area method. A single position mutation free energy profile was thus established and used as a scoring matrix to search peptides recognized by the Abl SH3 domain in the human genome. Our approach successfully picked ten out of 13 experimentally determined binding partners of the Abl SH3 domain among the top 600 candidates from the 218,540 decapeptides with the PXXP motif in the SWISS-PROT database. We expect that this physical-principle based method can be applied to other protein domains as well.
Conflict of interest statement
Figures
References
-
- Russell RB, Alber F, Aloy P, Davis FP, Korkin D, et al. A structural perspective on protein-protein interactions. Curr Opin Struct Biol. 2004;14:313–324. - PubMed
-
- Lim WA, Richards FM, Fox RO. Structural determinants of peptide-binding orientation and of sequence specificity in SH3 domains. Nature. 1994;372:375–379. - PubMed
-
- Feng SB, Chen JK, Yu H, Simon JA, Schreiber SL. Two binding orientations for peptides to the src SH3 domain: Development of a general model for SH3-ligand interactions. Science. 1994;266:1241–1247. - PubMed
-
- Macias MJ, Hyvonen M, Baraldi E, Schultz J, Sudol M, et al. Structure of the WW domain of a kinase-associated protein complexed with a proline-rich peptide. Nature. 1996;382:646–649. - PubMed
-
- Kuriyan J, Cowburn D. Modular peptide recognition domains in eukaryotic signaling. Annu Rev Biophys Biomol Struct. 1997;26:259–288. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
