

High-throughput DNA methylation profiling using universal bead arrays
- PMID: 16449502
- PMCID: PMC1415217
- DOI: 10.1101/gr.4410706
High-throughput DNA methylation profiling using universal bead arrays
Abstract
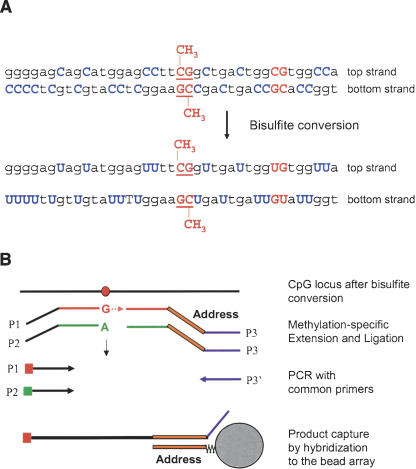
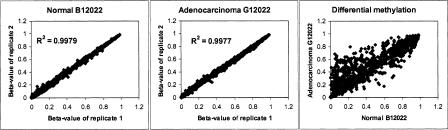
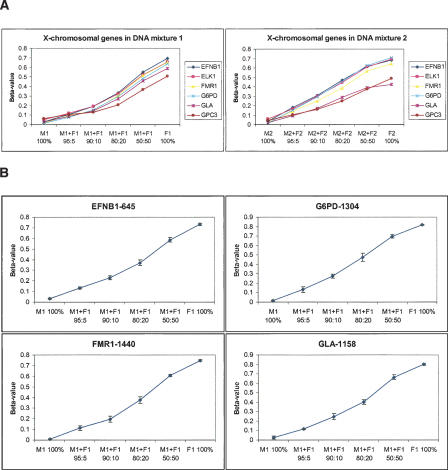
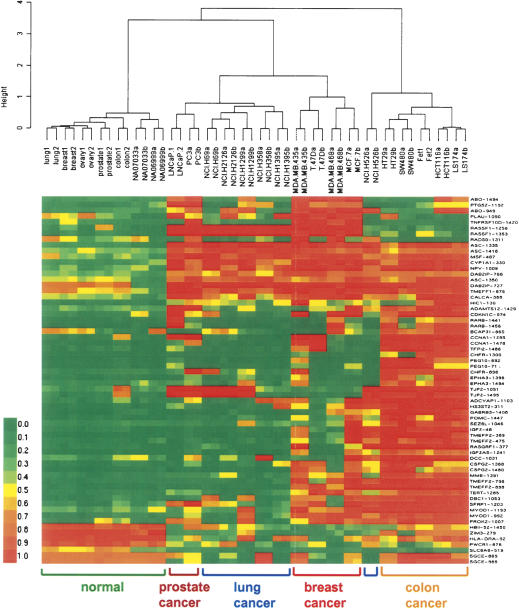
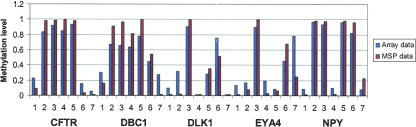
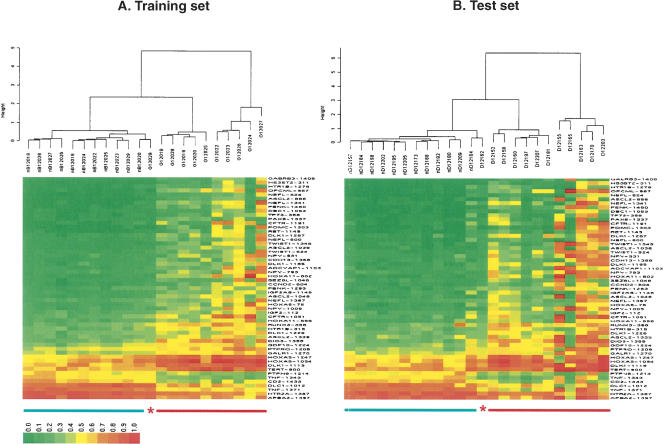
We have developed a high-throughput method for analyzing the methylation status of hundreds of preselected genes simultaneously and have applied it to the discovery of methylation signatures that distinguish normal from cancer tissue samples. Through an adaptation of the GoldenGate genotyping assay implemented on a BeadArray platform, the methylation state of 1536 specific CpG sites in 371 genes (one to nine CpG sites per gene) was measured in a single reaction by multiplexed genotyping of 200 ng of bisulfite-treated genomic DNA. The assay was used to obtain a quantitative measure of the methylation level at each CpG site. After validating the assay in cell lines and normal tissues, we analyzed a panel of lung cancer biopsy samples (N = 22) and identified a panel of methylation markers that distinguished lung adenocarcinomas from normal lung tissues with high specificity. These markers were validated in a second sample set (N = 24). These results demonstrate the effectiveness of the method for reliably profiling many CpG sites in parallel for the discovery of informative methylation markers. The technology should prove useful for DNA methylation analyses in large populations, with potential application to the classification and diagnosis of a broad range of cancers and other diseases.
Figures
References
-
- Akama, T.O., Okazaki, Y., Ito, M., Okuizumi, H., Konno, H., Muramatsu, M., Plass, C., Held, W.A., and Hayashizaki, Y. 1997. Restriction landmark genomic scanning (RLGS-M)-based genome-wide scanning of mouse liver tumors for alterations in DNA methylation status. Cancer Res. 57 3294–3299. - PubMed
-
- Alaminos, M., Davalos, V., Cheung, N.K., Gerald, W.L., and Esteller, M. 2004. Clustering of gene hypermethylation associated with clinical risk groups in neuroblastoma. J. Natl. Cancer Inst. 96 1208–1219. - PubMed
-
- Balmain, A., Gray, J., and Ponder, B. 2003. The genetics and genomics of cancer. Nat. Genet. 33 Suppl: 238–244. - PubMed
-
- Barker, D.L., Theriault, G., Che, D., Dickinson, T., Shen, R., and Kain, R. 2003. Self-assembled random arrays: High-performance imaging and genomics applications on a high-density microarray platform. Proc. SPIE 4966 1–11.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials