

A general model of G protein-coupled receptor sequences and its application to detect remote homologs
- PMID: 16452613
- PMCID: PMC2249772
- DOI: 10.1110/ps.051745906
A general model of G protein-coupled receptor sequences and its application to detect remote homologs
Abstract
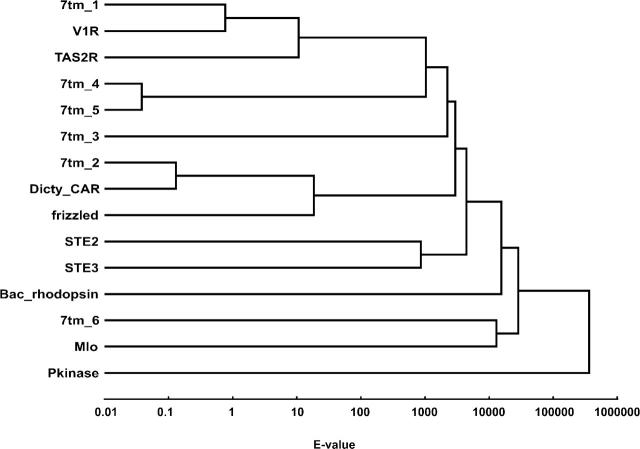
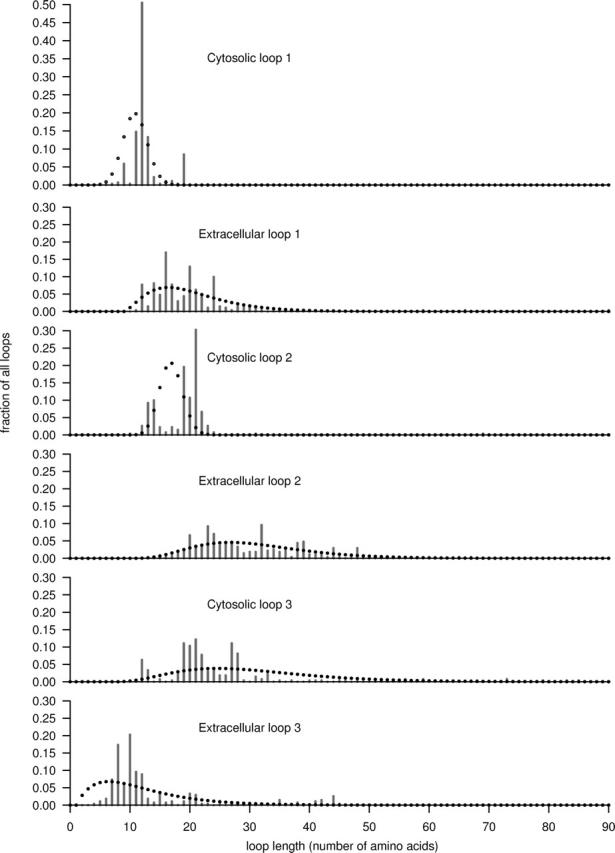
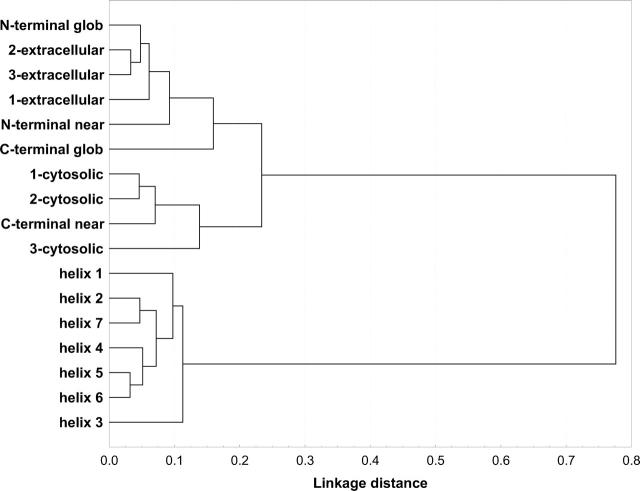
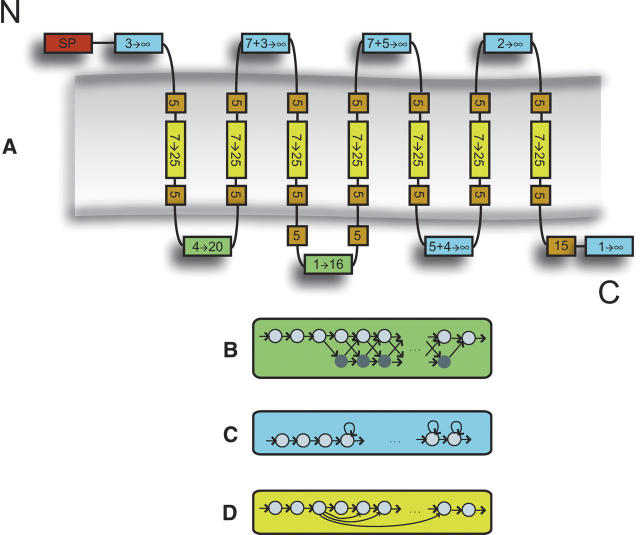
G protein-coupled receptors (GPCRs) constitute a large superfamily involved in various types of signal transduction pathways triggered by hormones, odorants, peptides, proteins, and other types of ligands. The superfamily is so diverse that many members lack sequence similarity, although they all span the cell membrane seven times with an extracellular N and a cytosolic C terminus. We analyzed a divergent set of GPCRs and found distinct loop length patterns and differences in amino acid composition between cytosolic loops, extracellular loops, and membrane regions. We configured GPCRHMM, a hidden Markov model, to fit those features and trained it on a large dataset representing the entire superfamily. GPCRHMM was benchmarked to profile HMMs and generic transmembrane detectors on sets of known GPCRs and non-GPCRs. In a cross-validation procedure, profile HMMs produced an error rate nearly twice as high as GPCRHMM. In a sensitivity-selectivity test, GPCRHMM's sensitivity was about 15% higher than that of the best transmembrane predictors, at comparable false positive rates. We used GPCRHMM to search for novel members of the GPCR superfamily in five proteomes. All in all we detected 120 sequences that lacked annotation and are potentially novel GPCRs. Out of those 102 were found in Caenorhabditis elegans, four in human, and seven in mouse. Many predictions (65) belonged to Pfam domains of unknown function. GPCRHMM strongly rejected a family of arthropod-specific odorant receptors believed to be GPCRs. A detailed analysis showed that these sequences are indeed very different from other GPCRs. GPCRHMM is available at http://gpcrhmm.cgb.ki.se.
Figures

Similar articles
-
A method for the prediction of GPCRs coupling specificity to G-proteins using refined profile Hidden Markov Models.BMC Bioinformatics. 2005 Apr 22;6:104. doi: 10.1186/1471-2105-6-104. BMC Bioinformatics. 2005. PMID: 15847681 Free PMC article.
-
Proteomic applications of automated GPCR classification.Proteomics. 2007 Aug;7(16):2800-14. doi: 10.1002/pmic.200700093. Proteomics. 2007. PMID: 17639603 Review.
-
Superfamily of G-protein coupled receptors (GPCRs)--extraordinary and outstanding success of evolution.Postepy Hig Med Dosw (Online). 2014 Oct 31;68:1225-37. doi: 10.5604/17322693.1127326. Postepy Hig Med Dosw (Online). 2014. PMID: 25380205 Review.
-
Predicting GPCR-G-protein coupling using hidden Markov models.Bioinformatics. 2004 Dec 12;20(18):3490-9. doi: 10.1093/bioinformatics/bth434. Epub 2004 Aug 5. Bioinformatics. 2004. PMID: 15297294
-
GPCR-MPredictor: multi-level prediction of G protein-coupled receptors using genetic ensemble.Amino Acids. 2012 May;42(5):1809-23. doi: 10.1007/s00726-011-0902-6. Epub 2011 Apr 20. Amino Acids. 2012. PMID: 21505826
Cited by
-
Prediction and expression analysis of G protein-coupled receptors in the laboratory stick insect, Carausius morosus.Turk J Biol. 2019 Feb 7;43(1):77-88. doi: 10.3906/biy-1809-27. eCollection 2019. Turk J Biol. 2019. PMID: 30930638 Free PMC article.
-
Whole proteome identification of plant candidate G-protein coupled receptors in Arabidopsis, rice, and poplar: computational prediction and in-vivo protein coupling.Genome Biol. 2008;9(7):R120. doi: 10.1186/gb-2008-9-7-r120. Epub 2008 Jul 31. Genome Biol. 2008. PMID: 18671868 Free PMC article.
-
No Evidence for Ionotropic Pheromone Transduction in the Hawkmoth Manduca sexta.PLoS One. 2016 Nov 9;11(11):e0166060. doi: 10.1371/journal.pone.0166060. eCollection 2016. PLoS One. 2016. PMID: 27829053 Free PMC article.
-
Controversy and consensus: noncanonical signaling mechanisms in the insect olfactory system.Curr Opin Neurobiol. 2009 Jun;19(3):284-92. doi: 10.1016/j.conb.2009.07.015. Epub 2009 Aug 5. Curr Opin Neurobiol. 2009. PMID: 19660933 Free PMC article. Review.
-
Expression and evolutionary divergence of the non-conventional olfactory receptor in four species of fig wasp associated with one species of fig.BMC Evol Biol. 2009 Feb 20;9:43. doi: 10.1186/1471-2148-9-43. BMC Evol Biol. 2009. PMID: 19232102 Free PMC article.
References
-
- Burge, C. and Karlin, S. 1997. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 268: 78–94. - PubMed
-
- Clyne, P.J., Warr, C.G., Freeman, M.R., Lessing, D., Kim, J., and Carlson, J.R. 1999. A novel family of divergent seven-transmembrane proteins: Candidate odorant receptors in Drosophila. Neuron 22: 327–338. - PubMed
-
- Clyne, P.J., Warr, C.G., and Carlson, J.R. 2000. Candidate taste receptors in Drosophila. Science 287: 1830–1834. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
