

Molecular models for the tissue specificity of DNA mismatch repair-deficient carcinogenesis
- PMID: 16464822
- PMCID: PMC1361617
- DOI: 10.1093/nar/gkj489
Molecular models for the tissue specificity of DNA mismatch repair-deficient carcinogenesis
Abstract
A common feature of all the known cancer genetic syndromes is that they predispose only to selective types of malignancy. However, many of the genes mutated in these syndromes are ubiquitously expressed, and influence seemingly universal processes such as DNA repair or cell cycle control. The tissue specificity of cancers that arise from malfunction of these apparently universal traits remains a key puzzle in cancer genetics. Mutations in DNA mismatch repair (MMR) genes cause the most common known cancer genetic syndrome, hereditary non-polyposis colorectal cancer, and the fundamental biology of MMR is one of the most intensively studied processes in laboratories all around the world. This review uses MMR as a model system to understand mechanisms that may explain the selective development of tumors in particular cell types despite the universal nature of this process. We evaluate recent data giving insights into the specific tumor types that are attributable to defective MMR in humans and mice under different modes of inheritance, and propose models that may explain the spectrum of cancer types observed.
Figures
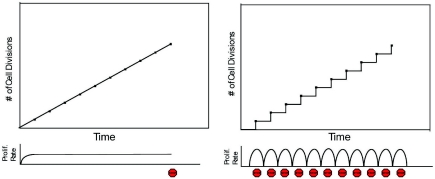

References
-
- Kolodner R.D., Marsischky G.T. Eukaryotic DNA mismatch repair. Curr. Opin. Genet. Dev. 1999;9:89–96. - PubMed
-
- Muller A., Fishel R. Mismatch repair and the hereditary non-polyposis colorectal cancer syndrome (HNPCC) Cancer Invest. 2002;20:102–109. - PubMed
-
- Fishel R., Lescoe M.K., Rao M.R., Copeland N.G., Jenkins N.A., Garber J., Kane M., Kolodner R. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer [Erratum (1994) Cell, 77, 167.] Cell. 1993;75:1027–1038. - PubMed
-
- Leach F.S., Nicolaides N.C., Papadopoulos N., Liu B., Jen J., Parsons R., Peltomaki P., Sistonen P., Aaltonen L.A., Nystrom-Lahti M., et al. Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer. Cell. 1993;75:1215–1225. - PubMed
