

tRNA properties help shape codon pair preferences in open reading frames
- PMID: 16473853
- PMCID: PMC1363775
- DOI: 10.1093/nar/gkj488
tRNA properties help shape codon pair preferences in open reading frames
Abstract
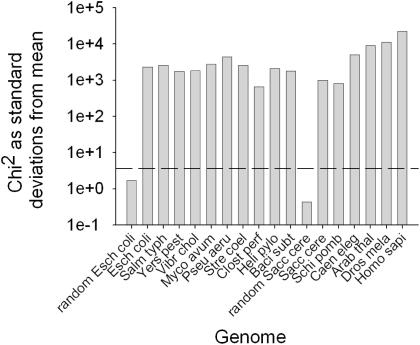
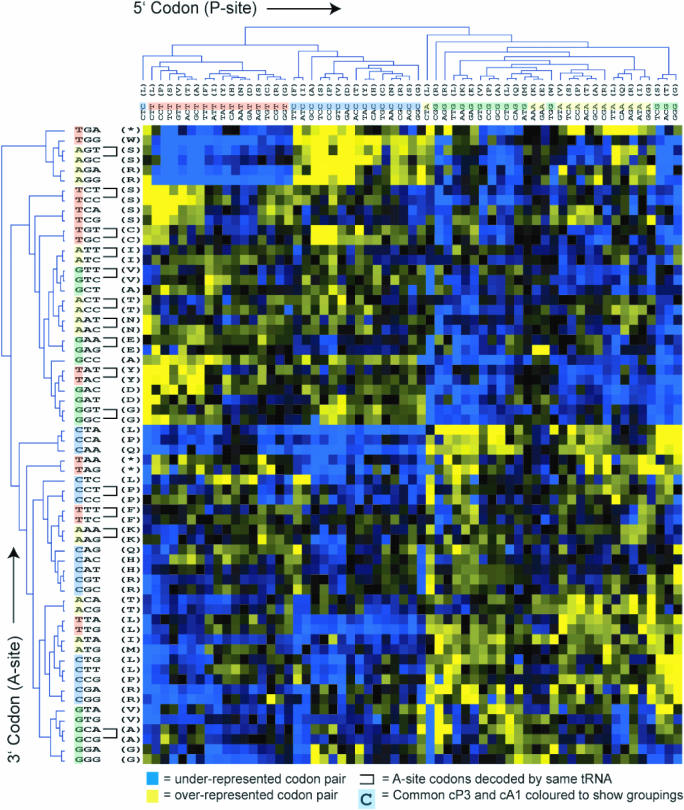
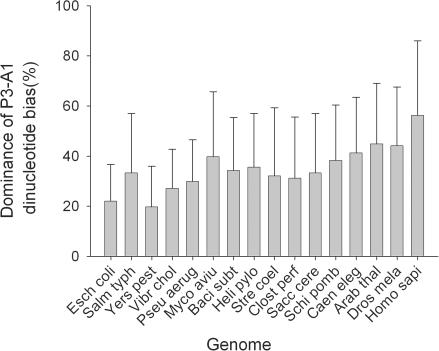
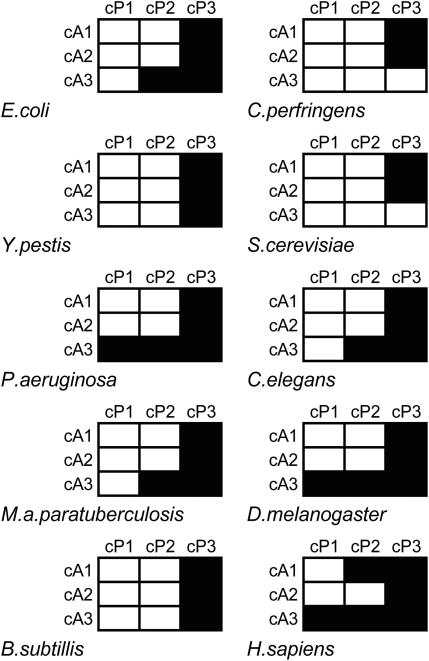
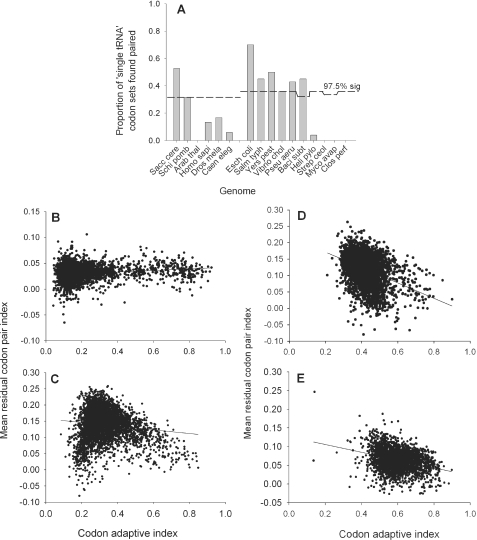
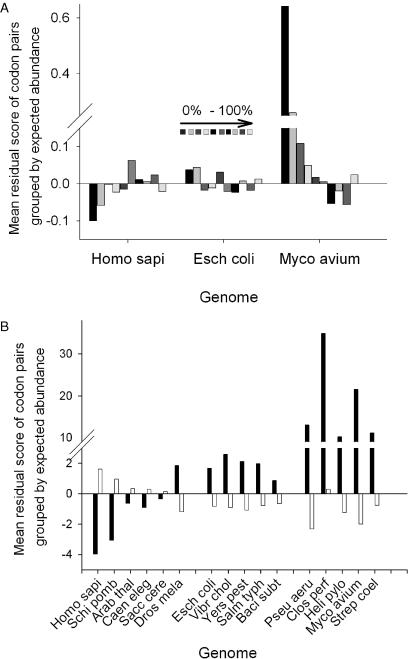
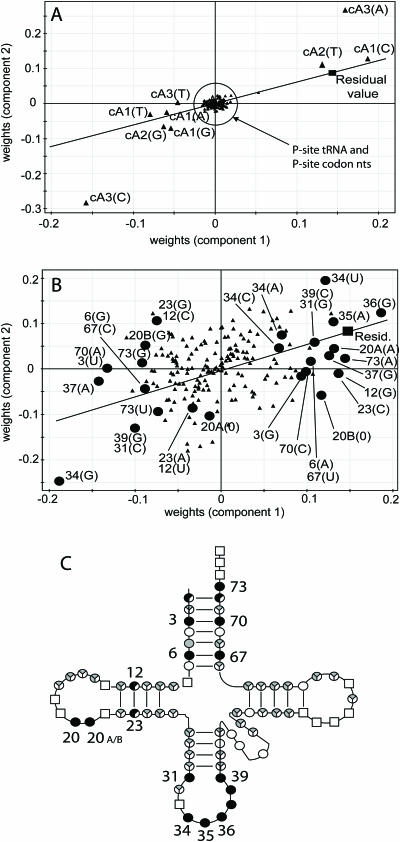
Translation elongation is an accurate and rapid process, dependent upon efficient juxtaposition of tRNAs in the ribosomal A- and P-sites. Here, we sought evidence of A- and P-site tRNA interaction by examining bias in codon pair choice within open reading frames from a range of genomes. Three distinct and marked effects were revealed once codon and dipeptide biases had been subtracted. First, in the majority of genomes, codon pair preference is primarily determined by a tetranucleotide combination of the third nucleotide of the P-site codon, and all 3 nt of the A-site codon. Second, pairs of rare codons are generally under-used in eukaryotes, but over-used in prokaryotes. Third, the analysis revealed a highly significant effect of tRNA-mediated selection on codon pairing in unicellular eukaryotes, Bacillus subtilis, and the gamma proteobacteria. This was evident because in these organisms, synonymous codons decoded in the A-site by the same tRNA exhibit significantly similar P-site pairing preferences. Codon pair preference is thus influenced by the identity of A-site tRNAs, in combination with the P-site codon third nucleotide. Multivariate analysis identified conserved nucleotide positions within A-site tRNA sequences that modulate codon pair preferences. Structural features that regulate tRNA geometry within the ribosome may govern genomic codon pair patterns, driving enhanced translational fidelity and/or rate.
Figures
References
-
- Maaloe O., Kjeldgaard N.O. Control of macromolecular synthesis. NY: W.A. Benjamin, Inc.; 1966.
-
- Kruger M.K., Pedersen S., Hagervall T.G., Sorensen M.A. The modification of the wobble base of tRNAGlu modulates the translation rate of glutamic acid codons in vivo. J. Mol. Biol. 1998;284:621–631. - PubMed
-
- Boe L. Translational errors as the cause of mutations in Escherichia coli. Mol. Gen. Genet. 1992;231:469–471. - PubMed
-
- Toth M.J., Murgola E.J., Schimmel P. Evidence for a unique first position codon-anticodon mismatch in vivo. J. Mol. Biol. 1988;201:451–454. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
