

Z-DNA-forming sequences generate large-scale deletions in mammalian cells
- PMID: 16473937
- PMCID: PMC1413824
- DOI: 10.1073/pnas.0511084103
Z-DNA-forming sequences generate large-scale deletions in mammalian cells
Abstract
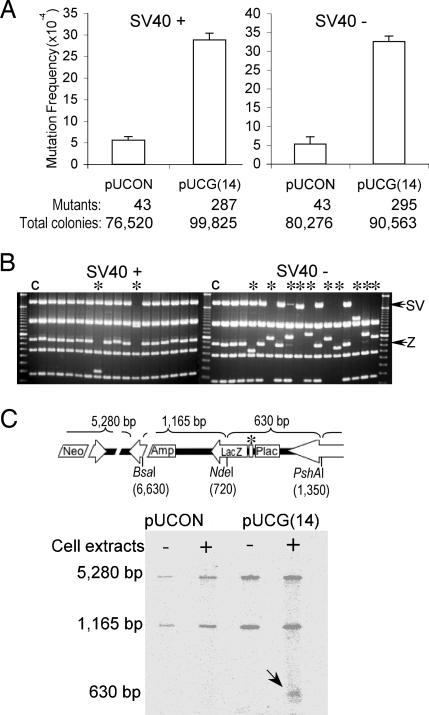
Spontaneous chromosomal breakages frequently occur at genomic hot spots in the absence of DNA damage and can result in translocation-related human disease. Chromosomal breakpoints are often mapped near purine-pyrimidine Z-DNA-forming sequences in human tumors. However, it is not known whether Z-DNA plays a role in the generation of these chromosomal breakages. Here, we show that Z-DNA-forming sequences induce high levels of genetic instability in both bacterial and mammalian cells. In mammalian cells, the Z-DNA-forming sequences induce double-strand breaks nearby, resulting in large-scale deletions in 95% of the mutants. These Z-DNA-induced double-strand breaks in mammalian cells are not confined to a specific sequence but rather are dispersed over a 400-bp region, consistent with chromosomal breakpoints in human diseases. This observation is in contrast to the mutations generated in Escherichia coli that are predominantly small deletions within the repeats. We found that the frequency of small deletions is increased by replication in mammalian cell extracts. Surprisingly, the large-scale deletions generated in mammalian cells are, at least in part, replication-independent and are likely initiated by repair processing cleavages surrounding the Z-DNA-forming sequence. These results reveal that mammalian cells process Z-DNA-forming sequences in a strikingly different fashion from that used by bacteria. Our data suggest that Z-DNA-forming sequences may be causative factors for gene translocations found in leukemias and lymphomas and that certain cellular conditions such as active transcription may increase the risk of Z-DNA-related genetic instability.
Conflict of interest statement
Conflict of interest statement: No conflicts declared.
Figures
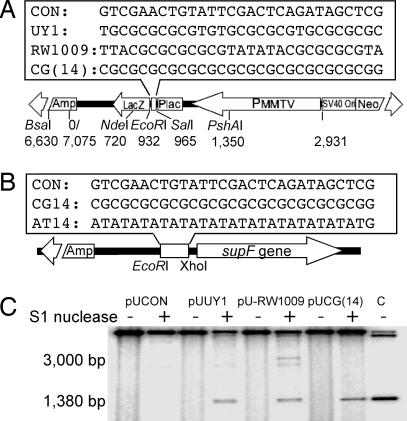
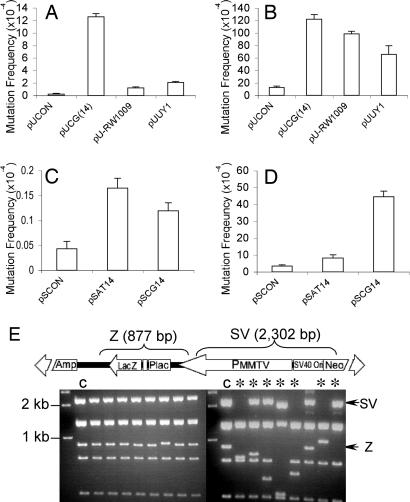
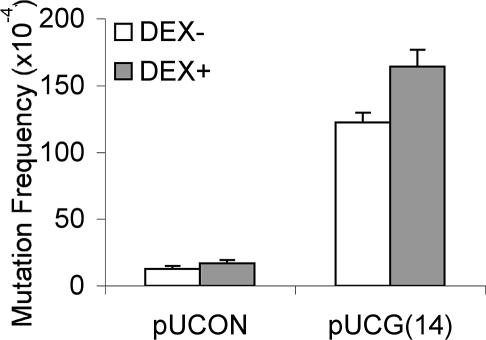
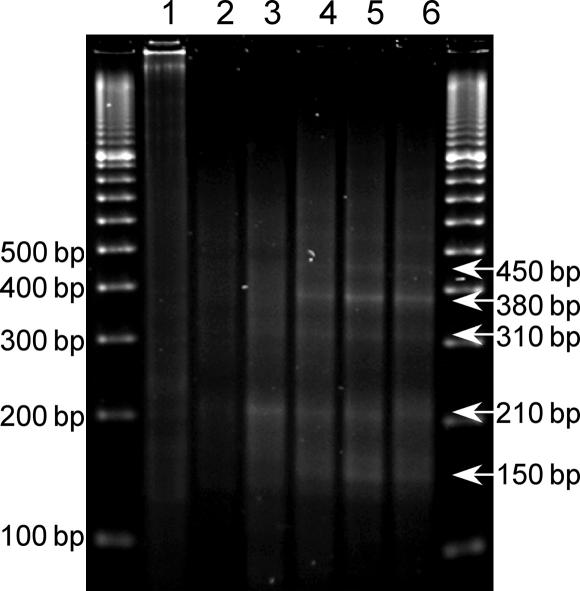
References
-
- Adachi M., Tsujimoto Y. Oncogene. 1990;5:1653–1657. - PubMed
-
- Seite P., Leroux D., Hillion J., Monteil M., Berger R., Mathieu-Mahul D., Larsen C. J. Genes Chromosomes Cancer. 1993;6:39–44. - PubMed
-
- Wolfl S., Wittig B., Rich A. Biochim. Biophys. Acta. 1995;1264:294–302. - PubMed
-
- Rimokh R., Rouault J. P., Wahbi K., Gadoux M., Lafage M., Archimbaud E., Charrin C., Gentilhomme O., Germain D., Samarut J., et al. Genes Chromosomes Cancer. 1991;3:24–36. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
