

Extensive recombination among human immunodeficiency virus type 1 quasispecies makes an important contribution to viral diversity in individual patients
- PMID: 16474154
- PMCID: PMC1395372
- DOI: 10.1128/JVI.80.5.2472-2482.2006
Extensive recombination among human immunodeficiency virus type 1 quasispecies makes an important contribution to viral diversity in individual patients
Abstract
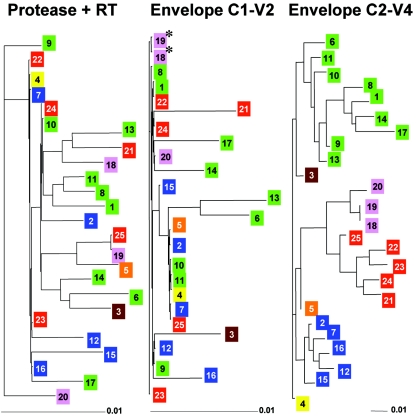
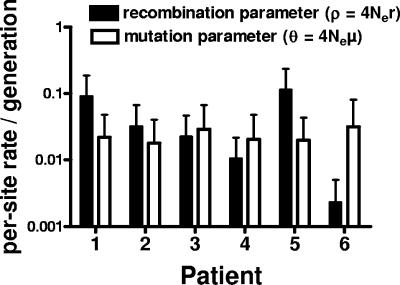
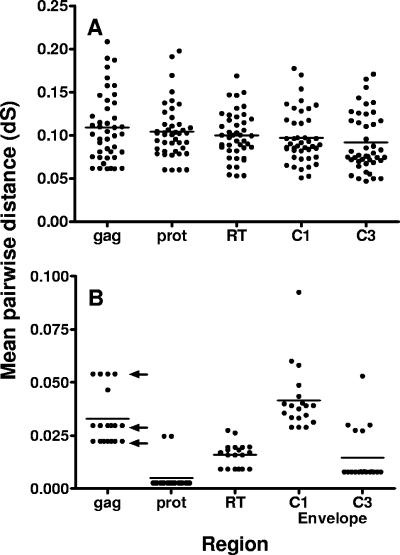
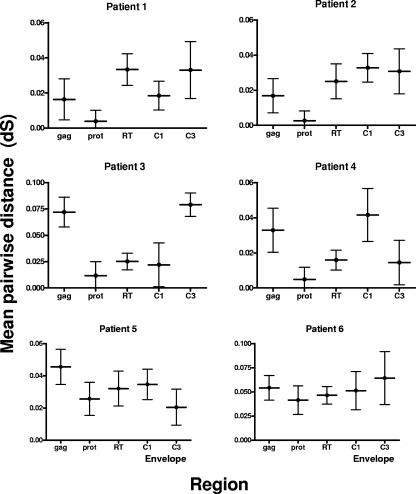
Although recombination during human immunodeficiency virus type 1 (HIV-1) replication in vitro and in vivo has been documented, little information is available concerning the extent that recombination contributes to the diversity of HIV-1 quasispecies in the course of infection in individual patents. To investigate the impact of recombination on viral diversity, we developed a technique that permits the isolation of contemporaneous clonal viral populations resulting from single infectious events by plasma-derived viruses, thereby permitting the assessment of recombination throughout the viral genomes, including widely separated loci, from individual patients. A comparison of the genomic sequences of clonal viruses from six patients, including patients failing treatment with antiretroviral therapy, demonstrated strong evidence for extensive recombination. Recombination increased viral diversity through two distinct mechanisms. First, evolutionary bottlenecks appeared to be restricted to minimal segments of the genome required to obtain selective advantage, thereby preserving diversity in adjacent regions. Second, recombination between adjacent gene segments appeared to generate diversity in both pol and env genes. Thus, the shuffling of resistance mutations within the genes coding for the protease and reverse transcriptase, as well as recombination between these regions, could increase the diversity of drug resistance genotypes. These findings demonstrate that recombination in HIV-1 contributes to the diversity of viral quasispecies by restricting evolutionary bottlenecks to gene segments and by generating novel genotypes in pol and env, supporting the idea that recombination may be critical to adaptive evolution of HIV in the face of constantly moving selective pressures, whether exerted by the immune system or antiretroviral therapy.
Figures
References
-
- Adzhubei, A. A., I. A. Adzhubei, I. A. Krasheninnikov, and S. Neidle. 1996. Non-random usage of ‘degenerate’ codons is related to protein three-dimensional structure. FEBS Lett. 399:78-82. - PubMed
-
- Bonhoeffer, S., C. Chappey, N. T. Parkin, J. M. Whitcomb, and C. J. Petropoulos. 2004. Evidence for positive epistasis in HIV-1. Science 306:1547-1550. - PubMed
-
- Brown, A. J., and A. Cleland. 1996. Independent evolution of the env and pol genes of HIV-1 during zidovudine therapy. AIDS 10:1067-1073. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
