

PhyloGibbs: a Gibbs sampling motif finder that incorporates phylogeny
- PMID: 16477324
- PMCID: PMC1309704
- DOI: 10.1371/journal.pcbi.0010067
PhyloGibbs: a Gibbs sampling motif finder that incorporates phylogeny
Abstract
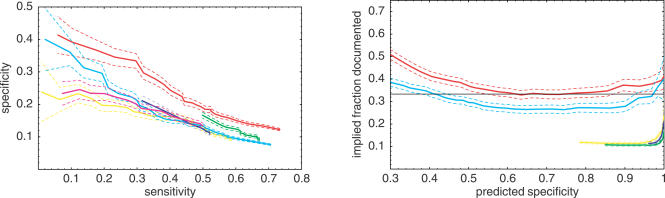
A central problem in the bioinformatics of gene regulation is to find the binding sites for regulatory proteins. One of the most promising approaches toward identifying these short and fuzzy sequence patterns is the comparative analysis of orthologous intergenic regions of related species. This analysis is complicated by various factors. First, one needs to take the phylogenetic relationship between the species into account in order to distinguish conservation that is due to the occurrence of functional sites from spurious conservation that is due to evolutionary proximity. Second, one has to deal with the complexities of multiple alignments of orthologous intergenic regions, and one has to consider the possibility that functional sites may occur outside of conserved segments. Here we present a new motif sampling algorithm, PhyloGibbs, that runs on arbitrary collections of multiple local sequence alignments of orthologous sequences. The algorithm searches over all ways in which an arbitrary number of binding sites for an arbitrary number of transcription factors (TFs) can be assigned to the multiple sequence alignments. These binding site configurations are scored by a Bayesian probabilistic model that treats aligned sequences by a model for the evolution of binding sites and "background" intergenic DNA. This model takes the phylogenetic relationship between the species in the alignment explicitly into account. The algorithm uses simulated annealing and Monte Carlo Markov-chain sampling to rigorously assign posterior probabilities to all the binding sites that it reports. In tests on synthetic data and real data from five Saccharomyces species our algorithm performs significantly better than four other motif-finding algorithms, including algorithms that also take phylogeny into account. Our results also show that, in contrast to the other algorithms, PhyloGibbs can make realistic estimates of the reliability of its predictions. Our tests suggest that, running on the five-species multiple alignment of a single gene's upstream region, PhyloGibbs on average recovers over 50% of all binding sites in S. cerevisiae at a specificity of about 50%, and 33% of all binding sites at a specificity of about 85%. We also tested PhyloGibbs on collections of multiple alignments of intergenic regions that were recently annotated, based on ChIP-on-chip data, to contain binding sites for the same TF. We compared PhyloGibbs's results with the previous analysis of these data using six other motif-finding algorithms. For 16 of 21 TFs for which all other motif-finding methods failed to find a significant motif, PhyloGibbs did recover a motif that matches the literature consensus. In 11 cases where there was disagreement in the results we compiled lists of known target genes from the literature, and found that running PhyloGibbs on their regulatory regions yielded a binding motif matching the literature consensus in all but one of the cases. Interestingly, these literature gene lists had little overlap with the targets annotated based on the ChIP-on-chip data. The PhyloGibbs code can be downloaded from http://www.biozentrum.unibas.ch/~nimwegen/cgi-bin/phylogibbs.cgi or http://www.imsc.res.in/~rsidd/phylogibbs. The full set of predicted sites from our tests on yeast are available at http://www.swissregulon.unibas.ch.
Conflict of interest statement
Figures

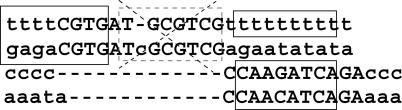
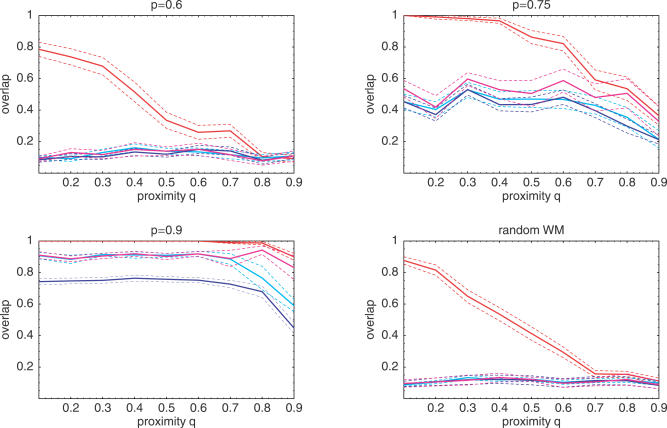
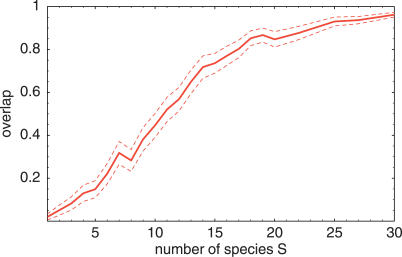
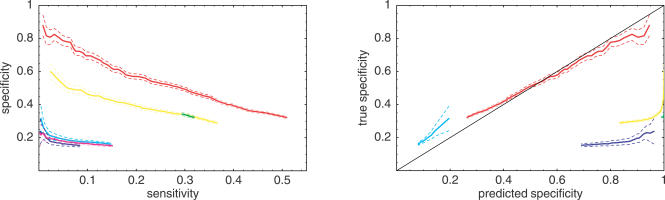
References
-
- Berg OG, von Hippel PH. Selection of DNA binding sites by regulatory proteins: Statistical-mechanical theory and application to operators and promoters. J Mol Biol. 1987;193:723–750. - PubMed
-
- Durbin R, Eddy S, Krogh G, Mitchison G. Cambridge University Press; 1998. Biological sequence analysis. 356 p.
-
- Lawrence CE, Altschul SF, Boguski MS, Liu JS, Neuwald AF, et al. Detecting subtle sequence signals: A Gibbs sampling strategy for multiple alignment. Science. 1993;262:208–214. - PubMed
-
- Liu JS, Neuwald AF, Lawrence CE. Markovian structures in biological sequence alignment. J Am Stat Assoc. 1999;94 :1–15.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
