

On the origin and highly likely completeness of single-domain protein structures
- PMID: 16478803
- PMCID: PMC1413790
- DOI: 10.1073/pnas.0509379103
On the origin and highly likely completeness of single-domain protein structures
Abstract
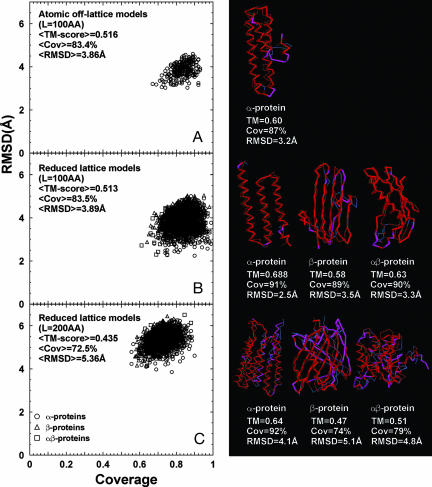
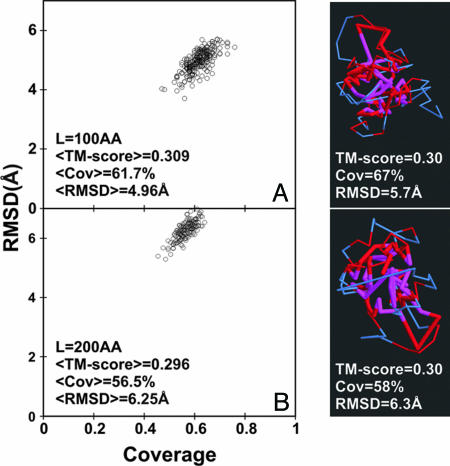
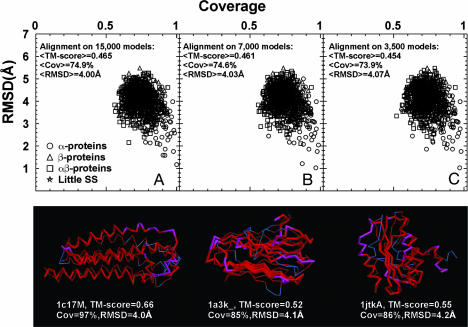
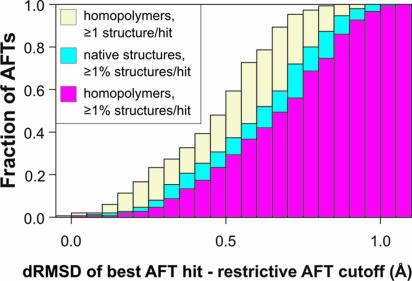
The size and origin of the protein fold universe is of fundamental and practical importance. Analyzing randomly generated, compact sticky homopolypeptide conformations constructed in generic simplified and all-atom protein models, all have similar folds in the library of solved structures, the Protein Data Bank, and conversely, all compact, single-domain protein structures in the Protein Data Bank have structural analogues in the compact model set. Thus, both sets are highly likely complete, with the protein fold universe arising from compact conformations of hydrogen-bonded, secondary structures. Because side chains are represented by their Cbeta atoms, these results also suggest that the observed protein folds are insensitive to the details of side-chain packing. Sequence specificity enters both in fine-tuning the structure and thermodynamically stabilizing a given fold with respect to the set of alternatives. Scanning the models against a three-dimensional active-site library, close geometric matches are frequently found. Thus, the presence of active-site-like geometries also seems to be a consequence of the packing of compact, secondary structural elements. These results have significant implications for the evolution of protein structure and function.
Conflict of interest statement
Conflict of interest statement: No conflicts declared.
Figures
References
-
- Anfinsen C. B. Science. 1973;181:223–230. - PubMed
-
- Todd A. E., Orengo C. A., Thornton J. M. Curr. Opin. Chem. Biol. 1999;3:548–556. - PubMed
-
- Card P. B., Gardner K. H. Methods Enzymol. 2005;394:3–16. - PubMed
-
- Chothia C., Finkelstein A. V. Annu. Rev. Biochem. 1990;59:1007–1039. - PubMed
-
- Burley S. K., Bonanno J. B. Annu. Rev. Genomics Hum. Genet. 2002;3:243–262. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
